
Bioinformatics Based Understanding of Effect of Mutations in the Human β Tubulin Outside Drug Binding Sites and its Significance in Drug Resistance
Abstract
Background:
Human β tubulin displays resistance to drugs like Taxol and Vinblastine due to amino acids substitutions within and outside the drug binding site.
Objective:
This study focuses on the effect of amino acid substitutions outside the drug binding site on drug resistance. Amino acid substitution like R306C (mut2) is associated with Taxol resistance and D197N (mut1) and K350N (mut3) are associated with Vinblastine resistance. However, the mechanism of resistance has not been understood yet. This study has attempted to investigate the mechanism of resistance.
Methods:
SWISSMODEL server was used to model the wild and the mutant β subunits which were later considered for protein-protein and protein-ligand docking using HADDOCK and AutoDock 1.5.6 software respectively. Dimer mutants were generated using Swisspdbviewer. POCASA 1.1 server was used to calculate the overall effect of substitution on pocket volume and the effect of substitution on domain mobility was explored using GROMACS software.
Results:
From sequence perspective, amino acid replacement in all three positions viz. D197N (mut1), R306C (mut2) and K350N (mut3) were found to have a deleterious effect on the stability of the protein. This study was further confirmed through structural analysis. Change in hydrogen bonding pattern was observed within the site of substitution in modeled mut1 and mut3 which is known to be specifically involved in Vinblastine interaction. In mut2 associated with Taxol binding, the hydrogen bonding pattern remained unaltered. All three mutants showed better protein-protein (β-β) interactions compared to the wild-type. Pocket size analysis in β subunit revealed that Taxol binding site increased in size after substitution in mut2 compared to the wild-type. However, the size of the Vinblastine binding site in the dimer interface remained the same before and after the substitution in wild and the mutants. Wild-type (β monomer and αβ dimer) associated with Taxol and Vinblastine, respectively showed better drug interaction compared to their mutants.
Conclusion:
This study throws light on the mechanism of drug resistance due to amino acid substitutions outside the drug binding site. It was found that amino acid substitution outside the drug site enhanced protein-protein interaction between the β-β subunits.
1. INTRODUCTION
Microtubules play a key role in cell division, mobility, architecture, intracellular transportation and chromosomal segregation [1]. The fundamental unit of a microtubule is a tubulin dimer made of α and β subunits [2]. Longitudinal interactions occur between β-α and α-β monomer of tubulin classified as intra- and inter-dimer interactions, respectively. As per literature review, intra-dimer interface (non-exchangeable) is rigid compared to the inter-dimer interface with exchangeable GDP [3, 4]. There are specific loop regions namely H1-S2 and M loops which exhibit lateral electrostatic interactions between H1-S2 loop of protofilament1 with M loop of adjacent protofilament2. Their overall binding affinity stabilizes the microtubule [4, 5].
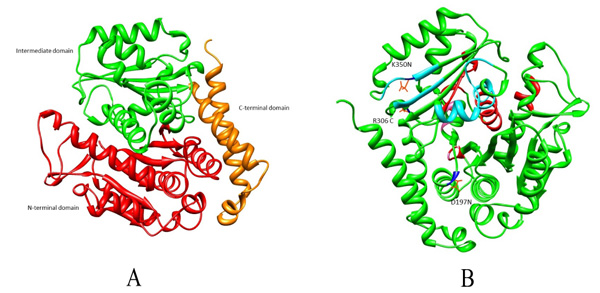
Microtubules are also considered as potential targets for anticancer drugs as their disruption results in cell cycle arrest associated with apoptosis [6]. Many anticancer drugs like Taxol, Vinblastine, Vincristine and Colchicine cause cell cycle arrest [7, 8]. It has been reported that amino acid substitutions have led to drug resistance in β tubulin. These substitutions could be within Taxol binding site (A231T, F270V, T274I, R282N and A364T) or outside the drug binding pockets (R306) [9, 10]. For Vinca alkaloid binding site, substitution within the binding pocket is S172A and substitution observed outside are D197N, K350N which causes altered drug binding affinity (Fig. 1). Given the high incidence of tubulin mutation as a cause of drug resistance in these studies, it is imperative to investigate the role of different amino acid substitutions in drug resistance [6, 9, 11-13]. Existing literature review investigated residues within the drug binding site [14-17]. However, not much has been reported for the substitutions outside the drug binding site. In the present study, the effect of these three mutations D197N, R306C and K350N outside the drug binding pocket and their role in drug resistance will be understood from docking and dynamics perspective.
2. MATERIALS AND METHODS
2.1. Amino Acid Composition Analysis
β tubulin of Homo sapiens was downloaded from Uniprot database (Accession number: Q9BVA1) [18]. To understand the effect of substitutions at positions 197, 306 and 350 we substituted each of them individually to generate D197N (mut1), R306C (mut2) and K350N (mut3). These mutants protein sequences were submitted to Amino Acid substitution (AAS) tools like PANTHER (Protein Analysis Through Evolutionary Relationships) [19] and Polyphen 2 (Polymorphism Phenotyping v2) [20] to study the effect of amino acid substitutions on structural stability. Consensus reports of these two softwares were considered for further analysis.
2.2. Homology Modeling and Mutant Generation
The protein sequence of human β tubulin was considered for homology modeling using SWISSMODEL server [21] to model the wild type. Of the listed templates, the 3D structure with better query coverage and amino acid identity was selected. Similar step was followed for modeling mut2 wherein Arg at 306th position was substituted by Cys. Regarding Vinblastine docking, instead of modeling and generating dimer, here we downloaded the 3D structure of Bos taurus (PDB id: 4I50) [22] from Protein Data Bank [23] which showed 99% amino acid identity with Homo sapiens. This has four subunits of which αβ dimer was extracted using Swisspdbviewer [21]. Furthermore, mut1 and mut3 were generated through the substitution of Asp and Lys with Asn at position 197 and 350, respectively. All the modelled structures were energy minimized using Swisspdbviewer software and were further validated using an PROSA (Protein Structure Analysis Software) [24].
2.3. Hydrogen Bonding Pattern
Hydrogen bonding patterns were separately investigated for the wild and the mutants using CHIMERA software [25].
2.4. Protein-Protein Docking
β subunit of the modeled wild and the mutants (mut1, mut2 and mut3) were considered for protein-protein docking (β-β) using HADDOCK server [26] to investigate their lateral binding affinity. The entire docking process consisted of three stages. They were (a) rigid-body energy minimization (b) a semi-flexible refinement torsion angle space and (c) a final refinement in explicit solvent. After each stage, structures were scored and ranked accordingly, and further selected for the next step. Moreover, HADDOCK score is the summation of van der Waals, electrostatic, desolvation and restraint violation energies with a buried surface area. From the webpage of HADDOCK, Easy interface module was selected to upload wild tubulins as molecule1 and molecule2. Regarding the active residues, sub-segments critically involved in lateral interactions like H1-S2 loop (residues 26-63) and M loop (residues 272-288) were considered for molecule1 and molecule2 respectively. Similar steps were followed for the mutant structures.
2.5. Pocket Identification
POCASA 1.1 software [27] was mainly used for measuring the ligand binding pocket size in monomer (for Taxol) and αβ dimer interface (for Vinblastine) of wild and mutants. Here, the probe radius and the grid size was 2 Å and 1.0Å respectively.
2.6. Protein-Ligand Docking
Modelled wild and the mutants were considered for docking using AUTODOCK tool 1.5.6 [28]. Drugs like Taxol and Vinblastine were obtained from 1JFF [29] and 1Z2B [30] which were separated from the receptor using Swisspdbviewer. Kollman charges were added to the modelled wild and the mutant receptors which were saved as .pdbqt files. Gasteiger charges were added to the ligands and saved as .pdbqt files. Firstly, wild type β subunit and Taxol were considered for docking. For the same, Gridbox was constructed near Taxol binding site (active site as reported in the crystal structure of 1JFF) wherein the number of grid points for x,y and z direction are 40,28 and 48, respectively. The grid centre for x,y and z coordinates are 6.855,-0.611 and -26.64, respectively. Same Gridbox value was considered for mut2 protein. Secondly, wild αβ dimer and Vinblastine were considered for docking. Basically, Vinca alkaloid prefers to bind to the dimer interface of the α-β subunits. The Gridbox is constructed near the inter-dimer interface (active sites as reported in the crystal structure of 1Z2B) wherein the number of grid point for x,y and z direction are 32,52 and 36, respectively. The grid centre for x, y and z coordinates are -0.413, 41.812 and 18.333, respectively. Same Gridbox values were considered for mut1 and mut3 proteins associated with Vinblastine docking. All docked poses were visualized using Discover Studio Visualizer software [31].
2.7. Protein Simulation
The coordinates of wild and mutants (monomer and dimer) were considered for molecular simulation using GROMACS 4.5.5 [32] with implementation of OPLS-AA/L force field [33]. These systems were solvated using TIP3P water modeling a cubic box with periodic boundary conditions [34]. Additionally, counter ions were added to neutralize the system. These systems were first energy minimized using steepest descent algorithm with a tolerance of 1000 kJ/mol/nm. Long-range electrostatic interactions were calculated using Particle Mesh Ewald (PME) summation with 1 nm cutoffs for Columbic interactions, and Van der Waal interactions were calculated with a distance cut-off of 1.4 nm. Later on, systems were equilibrated by applying positional restraints on the structure using NVT followed by NPT ensemble for 100 ps each. Temperature of 300 K was coupled using Berendsen thermostat [35] with pressure at 1 bar, coupled by Parrinello-Rahman algorithm [36]. The equilibrated systems were then subjected to 15 ns of production run with time-step integration of 2 fs. The trajectories were saved at every 2 ps and analyzed using analysis tools from GROMACS 4.5.5. Basic analyses like Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuations (RMSF) were calculated individually for wild and the mutants. RMSD measure the average distance between the atoms (usually the backbone atoms) of superimposed proteins [37]. The RMSF is one of the best methods of comparing dynamics. This calculation finds the degree of movement of each Cα around its average position, i.e. parts of the protein that are highly flexible will have a large RMSF value, while portions that are contained will result in a low RMSF [38].
3. RESULT
3.1. Amino Acid Substitution Analysis
Three β tubulin mutants mut1(D197N), mut2(R306C) and mut3(K350N) were generated through amino acid substitutions tools and as per their consensus report all three substitutions could probably damage the protein stability Table (1). However, this needs further confirmation from a structural perspective. For the same, the structures were modelled in our next step.
| Mutation | Polyphen | PANTHER | ||
|---|---|---|---|---|
| D197N | 0.99 | Probably damaging | 0.84 | -0.37 |
| R306C | 1 | Probably damaging | 0.9 | 0.1 |
| K350N | 0.99 | Probably damaging | 0.79 | -0.75 |
3.2. Homology Modeling and Mutant Generation
SWISSMODEL server was considered for the modeling of human wild β subunit and mut2 which was later considered for Taxol binding. This model was based on the template of 4I50 of Bos Taurus with query coverage of 99%. For Vinblastine docking, αβ dimer itself was extracted from PDB id 4I50. Both wild and the mutant models showcased good local model quality below 0 for a window size of 40 residues (Supplementary material-S1 (a-d)).
3.3. Hydrogen Bonding Pattern
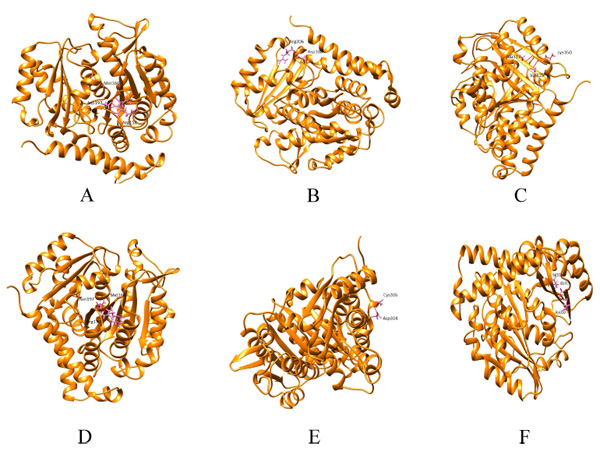
Hydrogen bonding patterns revealed that in wild β subunit, salt bridges were observed between Asp197 and Arg158 of helix4 wherein OD1 and OD2 of Asp197 interacted with the hydrogen atom of Arg158. This interaction is basically associated with the anchoring of COOH-terminal end of helix 4 during lateral protofilament interaction as per literature review [39]. Furthermore, hydrogen atom of Asp197 (renumbered as 199 in wild-type after modeling) interacts with the oxygen and hydrogen atom of Met166 and Arg158. Thus, in total, there are five interactions observed between Asp199 and the other two amino acids (Met166, Arg158). After the substitution, mut1 displays only three hydrogen bonds between Met166 and Arg158. In Arg306 (wild) and mut2 the hydrogen bonding pattern remains the same. With respect to Lys350 (wild), the hydrogen bonds were observed between Thr353 and Ala317. However, in mut3 Asn350 interacts with Ala315 and Val313. With substitution, the contact with Thr353 ceases. Overall, mut1 and mut3 showcases fewer hydrogen bonds which are otherwise unscathed in mut2 (Fig. 2a-f).
3.4. Protein-Protein Docking
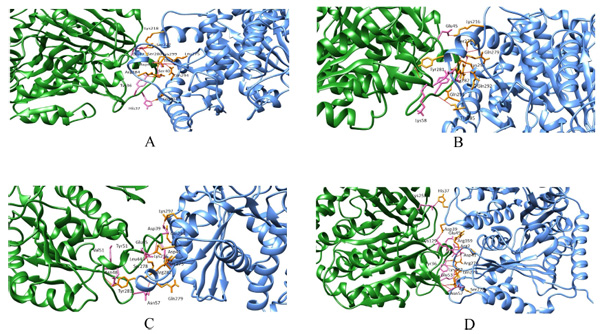
Wild and the mutant models were considered for protein-protein docking to calculate their overall binding affinity and the protofilament interactions before and after mutations. In wild beta subunits, the binding affinity score is -76.9 +/-8.4 with a buried surface of 1643.5 +/-64.4. Conversely, the binding affinity of mut1, mut2 and mut3 were -112.6 +/-4.0,-106.0 +/-19.1,-92 +/-4.2, respectively. Moreover, the buried surface of mut1, mut2 and mut3 were 1685.4 +/-96.4, 2108.3 +/-155.5 and 1607.0 +/-57.3 respectively. Thus, here a considerable increase in the binding affinity between mut1, mut2 and mut3 was observed compared to their wild counterpart. Similarly, a steady rise in the buried surface area was observed in mut1 and mut2 with an exception in mut3 (Fig. 3a-d). Thus, mutants showed better lateral interactions which could assist in better protofilament interactions during microtubule formation.
3.5. Pocket Identification
As per the POCASA report, in wildtype β subunit the pocket size associated with Taxol interaction is 242Å3 which increases to 251Å3 in mut2. Thus, an increase in pocket size was observed after the substitution of arginine with cysteine. In Vinblastine binding dimer interface, the wild and the mutant dimmers displayed a pocket size of 268Å3 which remained unaltered after the substitution in D197N and K350N.
3.6. Protein-Ligand Docking
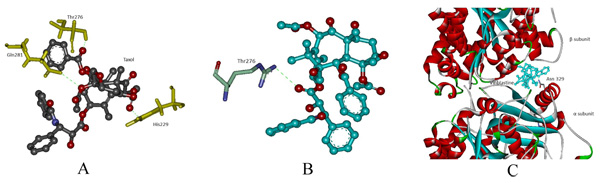
In protein-ligand docking, drug Taxol was docked with modeled wild and mut2. In wild type, the binding affinity is -8.1 kcal/mol wherein Taxol interacts with His229, Thr276 and Gln281. In mut2, the binding affinity is –6.4 kcal/mol and a single interaction with Thr276 was observed after the substitution of Arg with Cys. Here, we assume that an increase in pocket size volume after substitution could be the reason behind the absence two interactions (Figs. 4a, b). Furthermore, Vinblastine was docked with wild and the generated mutant dimers. In wild-type, the binding affinity is -7.9 kcal/mol and the drug interacts with Asn 329. In mut1 and mut3 the binding affinity is -7.4 kcal/mol and -6.8 kcal/mol respectively with no interaction between the receptor and the drug. The overall change in hydrogen bonding pattern within the site of substitution could be the reason behind the reduced binding affinity (Fig. 4c).
3.7. Molecular Simulation
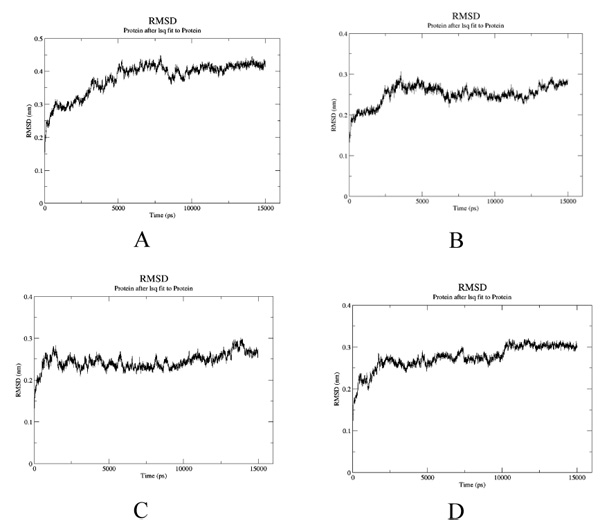
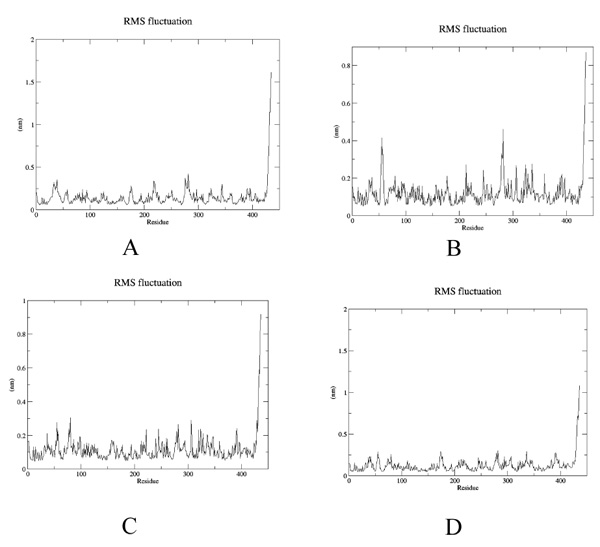
To estimate the effect of mutation on structural stability/flexibility of the tubulins with respect to the wild type, the RMSD and RMSF values were calculated individually. Here, we monitored the structural stability of the wild and the mutants throughout the simulation (Fig. 5a-d). In mut1, mut2 and mut3 there is a dramatic increase in the RMSD compared to the wild type. As per the RMSF report, in mut1 and mut2 the subsegment of D197N and R306C are flexible compared to the wild type. However, in mut3 the subsegment K350N shows an increase in rigidness compared to the wild type after substitution with asparagine (Fig. 6a-d).
4. DISCUSSION
It is generally observed that substitutions within the drug binding site of β tubulin lead to resistance. Since there is a paucity of information about substitution outside the drug binding site, in the present study the role of amino acid substitutions outside the drug binding site in β tubulin from Homo sapiens has been investigated. Firstly, from sequence perspective, all three substitutions (D197N, R306C and K350N) were found to be probably damaging the protein stability. Secondly, hydrogen bonding network analysis of the wild and the mutant structures reveals a decrease in bonds in mut1 and mut3 compared to the wild-type. In particular, salt bridge formation between Asp197 and Arg158 is lost in mut1 after substitution which increases the flexibility of this region as observed during simulation. Mut2 also shows higher flexibility irrespective of no change in their bonding pattern. In mut3, substitution reduces the flexibility of this subsegment. Thirdly, protein-protein interaction analysis through docking confirms better interactions among mut1-mut1, mut2-mut2 and mut3-mut3 compared to the wildtype. Thus, a better protofilament interaction is inevitable after amino acid substitutions which could further enhance their domain stability. Fourthly, protein-ligand docking analysis reveals lower Vinblastine binding affinity in mut1 and mut3 compared to the wild-type due to the loss of salt bridge formation and higher domain flexibility which could ultimately reduce the protein-ligand interaction. Even in mut2, higher flexibility associated with larger pocket size ultimately hampered with the Taxol binding. This was evident from a single residue binding instead of three. In mut3, the domain rigidness plays a crucial role in reduced Vinblastine binding within the pocket. Thus, all three mutants have reduced drug binding and improved protein-protein interactions after substitutions which could certainly assist in better protofilament interactions much needed for their stability. Thus to conclude, even mutations outside the drug binding site have critical role in protein-protein and protein–ligand interaction which may play pivotal role during drug resistance.
CONCLUSION
In this study, we investigated the effect of amino acid substitutions outside the drug binding site on drug resistance. And it was observed that amino acid substitution outside the drug site enhanced protein-protein interaction between the β-β subunits.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Website along with the published article.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
No Animals/Humans were used for studies that are base of this research.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Authors would like to acknowledge the Department of Biotechnology (DBT), Government of India, sponsored Distributed Information Sub Centre (SubDIC) of Biotechnology Information System (BTIS) Network at ACTREC where analysis of MD trajectories and docking studies were carried out. Authors also thank Super-computing facility (Utkarsh) of Computer division, BARC, for providing access to molecular dynamics simulation of structures. This Project was not funded by any external funding agencies.

