
iMPT-FRAKEL: A Simple Multi-label Web-server that Only Uses Fingerprints to Identify which Metabolic Pathway Types Compounds can Participate In
Abstract
Background:
Metabolic pathway is one of the most basic biological pathways in living organisms. It consists of a series of chemical reactions and provides the necessary molecules and energies for organisms. To date, lots of metabolic pathways have been detected. However, there still exist hidden participants (compounds and enzymes) for some metabolic pathways due to the complexity and diversity of metabolic pathways. It is necessary to develop quick, reliable, and non-animal-involved prediction model to recognize metabolic pathways for any compound.
Methods:
In this study, a multi-label classifier, namely iMPT-FRAKEL, was developed for identifying which metabolic pathway types that compounds can participate in. Compounds and 12 metabolic pathway types were retrieved from KEGG. Each compound was represented by its fingerprints, which was the most widely used form for representing compounds and can be extracted from its SMILES format. A popular multi-label classification scheme, Random k-Labelsets (RAKEL) algorithm, was adopted to build the classifier. Classic machine learning algorithm, Support Vector Machine (SVM) with RBF kernel, was selected as the basic classification algorithm. Ten-fold cross-validation was used to evaluate the performance of the iMPT-FRAKEL. In addition, a web-server version of such classifier was set up, which can be assessed at http://cie.shmtu.edu.cn/impt/index.
Results:
iMPT-FRAKEL yielded the accuracy of 0.804, exact match of 0.745 and hamming loss of 0.039. Comparison results indicated that such classifier was superior to other models, including models with Binary Relevance (BR) or other classification algorithms.
Conclusion:
The proposed classifier employed limited prior knowledge of compounds but gives satisfying performance for recognizing metabolic pathways of compounds.
1. INTRODUCTION
Metabolomics is an important part of systems biology. Many life activities in cells occur at the metabolite level, such as cell signaling, energy transfer, and cell-to-cell communication. At present, metabolomics has developed rapidly and penetrated into many fields, including disease diagnosis, pharmaceutical research and development, nutritional food science, toxicology, environmental science, and botany, which are highly related to human health care. Metabolomics includes several metabolic pathways, and each metabolic pathway is composed of a series of continuous chemical reactions. Each reaction is catalyzed by an enzyme and changes from one molecule to another and provides cells necessary molecules and energy to sustain the life of the organism [1]. Thus, metabolic pathway is one of the most basic pathways in living organisms. A good understanding of metabolic pathways is very helpful for studying the mechanisms of some basic biological processes.
In the past ten years, lots of metabolic pathways have been detected for many organisms, and this information is stored in online public databases. Kyoto Gene and Genome Encyclopedia (KEGG) [2, 3] database are one of the most popular metabolome databases, including metabolic pathways and interaction network information. In KEGG PATHWAY (https://www.genome.jp/kegg/pathway.html), metabolic pathways are classified into 12 types: (1) Carbohydrate metabolism; (2) Energy metabolism; (3) Lipid metabolism; (4) Nucleotide metabolism; (5) Amino acid metabolism; (6) Metabolism of other amino acids; (7) Glycan biosynthesis and metabolism; (8) Metabolism of cofactors and vitamins; (9) Metabolism of terpenoids and polyketides; (10) Biosynthesis of other secondary metabolites; (11) Xenobiotics biodegradation and metabolism; (12) Chemical structure transformation maps. As mentioned above, the compounds are the main component for each metabolic pathway. It is essential to correctly predict which metabolic pathway types a compound can participate in. Such study is helpful to find out new participants for an existing metabolic pathway. Clearly, such prediction via traditional experiments is of low efficiency and high cost. Developing effective computational methods is an alternative way.
To date, several computational methods have been proposed in this regard. The first work was proposed by Cai et al. [4] Their method used functional groups to represent each compound and adopted the Nearest Neighbor Algorithm (NNA) [5] as the prediction engine. Later, Lu et al. proposed a more powerful method, which employed AdaBoost as the classification algorithm [1]. However, these two studies only considered compounds belonging to the exact one pathway type. In fact, several compounds can participate in more than one pathway types. Thus, several investigators followed by developing multi-label classifiers. In 2011, Hu et al. proposed a multi-label classifier with the chemical-chemical interaction information in STITCH [6]. Gao et al. fused the interactions of chemicals and proteins to build a classifier with wide applications because this method can not only assign compounds to metabolic pathway types but also predict the metabolic pathway types of enzymes, another main component of the metabolic pathway [7]. Chen et al. [8] used the minimum redundant maximum correlation (mRMR) [9] method to analyze molecular fragment features of compounds, thereby selecting optimal features to build the multi-label classifier with the help of Support Vector Machine (SVM) [10]. The above-mentioned classifiers only output the rank of metabolic pathway types for a given compound; that is, they cannot determine which types were predictions. Recently, the other two methods were proposed. Fang and Chen converted the original multi-label classification problem into a binary classification problem by pairing compounds and pathway types as samples [11]. However, the section of negative samples is a problem; different negative samples can induce different models. Guo et al. built a binary classifier for each pathway type with a complex compound representation scheme and SVM [12]. This method constructed a classifier for each pathway type. For a given compound, users have to execute several classifiers to determine its pathway types, increasing the computational complex.
To partly overcome the defects of the above-mentioned methods and build a new multi-label classifier with wide applications, we used the most classic and widely used form, fingerprints, to represent each compound, which can be extracted from its Simplified Molecular Input Line Entry System (SMILES) [13] format. Then, the Random k-labelsets (RAKEL) algorithm [14, 15] was adopted to process the multi-label problem. The SVM with RBF kernel was adopted to build basic classifiers, thereby constructing a multi-label classifier, namely iMPT-FRAKEL. The proposed classifier can determine specific metabolic pathway types for a given compound rather than only giving a pathway type rank, as reported in previous studies [6-8]. On the other hand, the construction procedures of iMPT-FRAKEL were not involved in negative sample selection, overcoming the problem in a study [11], and it is a unified model for predicting the metabolic pathway types of compounds, improving the method in another study [12] that consisted of several classifiers. Furthermore, the proposed classifier used limited prior knowledge of compounds because it can make prediction as long as the SMILES format of compounds were available. Thus, our classifier had wider applications than most previous classifiers, which always needed several prior knowledge of compound, such as chemical interaction information. The ten-fold cross-validation on iMPT-FRAKEL indicated that the accuracy and exact match were 0.804 and 0.745, respectively, suggesting high performance of the classifier. In addition, a web-server with the same name was developed, which can be accessed at http://cie.shmtu.edu.cn/impt/index.
2. MATERIALS AND METHODS
2.1. Benchmark Dataset
Details of the metabolic pathways were obtained from the KEGG PATHWAY (http://www.kegg.jp/kegg/pathway.html) (accessed in September 2019) [2, 3]. 5,641 compounds that can participate in at least one metabolic pathway were obtained. After excluding compounds without representations of SMILES [13] and ECFP [16] fingerprints, we finally obtained 4,739 compounds. The detailed information of these compounds can be accessed at http://cie.shmtu.edu.cn/impt /index. As mentioned in Section 1, metabolic pathways in KEGG are classified into 12 types. Accordingly, 4,739 compounds can also be classified into 12 classes in a way that if a compound belongs to a pathway that is in one pathway type, such compound is assigned to this pathway type. For an easy description, we tagged 12 pathway types as P1, P2, ..., and P12, respectively. The correspondence of pathway type names and these tags is shown in columns 1 and 2 of Table 1. The number of compounds in each pathway is also listed in this table. The total number of compounds in 12 pathway types were 5,784, which was larger than the total number of different compounds (4,739), suggesting that some compounds belonged to more than one metabolic pathway. Thus, it is a typical multi-label classification problem for assigning compounds to pathway types.
2.2. Representation of Compounds
To construct an efficient classifier, each sample should be encoded into a series of numbers, which contains essential properties of samples. In cheminformatics, SMILES [13] is the most classic and widely used scheme for representing compounds [17-20]. By this scheme, each compound was represented by a line notation with ASCII strings. Then, fingerprints can be extracted from this representation, which were collected in a binary vector. In this study, we first obtained the SMILES format of 4,739 compounds from STITCH and used RDKit [21] to access ECFP [16] fingerprints of each compound. Obtained binary vectors for investigated compounds are available at http://cie.shmtu.edu.cn/impt/index.
| Tag | Metabolic Pathway Type | Number of Compounds |
|---|---|---|
| P1 | Carbohydrate metabolism | 448 |
| P2 | Energy metabolism | 174 |
| P3 | Lipid metabolism | 512 |
| P4 | Nucleotide metabolism | 149 |
| P5 | Amino acid metabolism | 553 |
| P6 | Metabolism of other amino acids | 203 |
| P7 | Glycan biosynthesis and metabolism | 70 |
| P8 | Metabolism of cofactors and vitamins | 413 |
| P9 | Metabolism of terpenoids and polyketides | 866 |
| P10 | Biosynthesis of other secondary metabolites | 925 |
| P11 | Xenobiotics biodegradation and metabolism | 932 |
| P12 | Chemical structure transformation maps | 539 |
| Total number of compounds | 5,784 | |
| Total number of different compounds | 4,739 | |
2.3. Multi-label Classification Model
As described in Section 2.1, some compounds had multiple pathway types, inducing a multi-label classification problem. Generally, there are two ways for building multi-label classification models: (1) problem transformation; (2) algorithm adaption. The first one converts the original problem into several single-label classification models, while the second one directly reforms the specific single-label classification algorithm such that it can tackle multi-label classification problems. In this study, we adopted the first way to construct the model. The well-known method, RAKEL algorithm [14, 15], was employed, which has been applied to deal with several biological problems [20, 22-27].
The RAKEL algorithm extends another multi-label classification method, Label Powerset (LP) algorithm [28, 29], which treats a combination of labels as a new label, thereby converting into a single-label classification problem. However, this algorithm has several defects, such as high computational cost, sample skew, etc. In view of this, Tsoumakas et al. proposed the RAKEL algorithm [14, 15]. It breaks the initial set of labels into m label subsets with small size k. On each label subset, LP algorithm is adopted to train a multi-label classifier, namely LP classifier. For example, given a label subset {l1,l2,...,lk}, its power set is defined as the new label set. These new labels are assigned to each sample according to its original labels. Accordingly, each sample has only one new label. A LP classifier is constructed on the dataset with new labels based on a given classification algorithm. The model built by RAKEL algorithm always contains m constructed LP classifiers. For an input sample s, each LP classifier gives its binary decision on each involved label. For each label, RAKEL algorithm counts the average of the binary decisions yielded by LP classifiers, whose underlying label set contains such label. If the average is larger than a predefined threshold, which is always set to 0.5, the label is assigned to the input sample. As mentioned above, there are two main parameters for RAKEL algorithm, m and k, where k determines the size of label subset and m stands for the number of label subsets or the number of LP classifiers. On the other hand, the basic single-label classification algorithm is also an important factor to build effective RAKEL classifiers. The detailed descriptions of RAKEL algorithm can be obtained from another study [15].
To quickly implement RAKEL algorithm, Meka (http://waikato.github.io/meka/) [30] was employed, which is an open-source machine learning framework collecting several multi-label classification scheme. One tool, named ‘RAKEL’, implements RAKEL algorithm. We tried several values of m and k and selected the best ones to construct the final classifier. Furthermore, two classic single-label classification algorithms: SVM [10] and random forest (RF) [31], were tried. For easy descriptions, models constructed by RAKEL algorithm were called RAKEL models.
2.4. Classification Algorithm
To construct LP classifiers, one single-label classification algorithm was necessary. Here, we tried two classic classification algorithms, SVM [10] and RF [31], and we finally selected the best one. Their brief descriptions were as below.
The principle of SVM is to select an appropriate kernel (such as RBF kernel) to map all samples in the training data set to a higher-dimensional space, in which samples in different classes can easily be separated by a hyperplane. Given a kernel, the training procedure of SVM is to find out an optimal hyperplane. For a query sample, its class is determined according to which side of hyperplane it belongs to. To date, several types of SVM have been proposed to deal with different problems and they have wide applications in bioinformatics [20, 22, 32-36]. In this study, we used the SVM whose training procedures were optimized by Sequential Minimal Optimization (SMO) algorithm [37]. The kernel was set to polynomial kernel or RBF kernel.
RF [31] is another widely used classification algorithm. It always consists of several decision trees. Each tree was built with samples randomly selected, with replacement, from the original training dataset and randomly selected features. Although decision tree is a weak classification algorithm, RF is a relative much more powerful algorithm [38]. Thus, RF is always an important choice to construct classifiers in the fields of bioinformatics and computational biology [17, 18, 39-44]. The number of decision trees is the most important parameter for RF. We tried several values for this parameter in this study.
All the above-mentioned SVM and RF have been integrated in Meka [30]. They were directly invoked in the tool ‘RAKEL’.
2.5. Construction of iMPT-FRAKEL
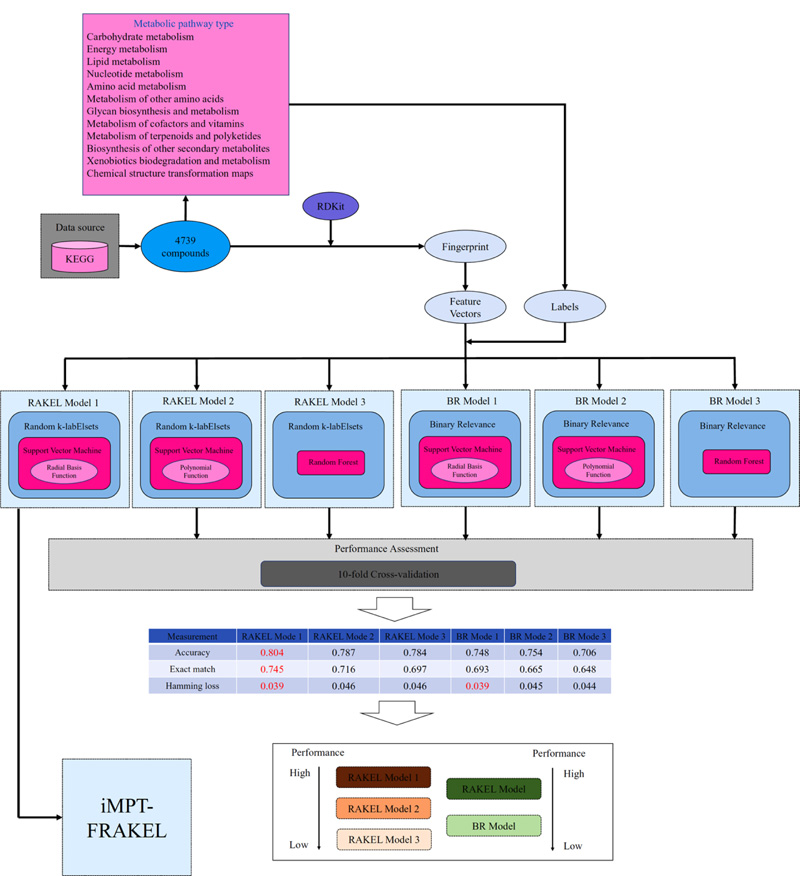
According to the dataset and methods mentioned above, we constructed a multi-label classifier, named iMPT-FRAKEL, for prediction of the metabolic pathway types of compounds. The entire procedures are illustrated in Fig. (1). First, 4739 compounds were retrieved from KEGG PATHWAY and constituted the underlying dataset. Then, each compound was converted into its SMILES format, from which its fingerprints were extracted via RDKit. These fingerprints were encoded into a 1024-D vector. Based on the pathway types of each compound, we assigned these pathway type labels to the corresponding vector. The vectors together with their labels were fed into the RAKEL algorithm, which incorporated SVM with RBF kernel as the classification algorithm, to construct iMPT-FRAKEL.
2.6. Assessment and Measurement
To evaluate the performance of each classifier in this study, ten-fold cross validation [45] was used. This method divides the original training samples randomly and equally into ten subsets. Samples in each subset are singled out one by one as testing samples, while samples in the remaining nine subsets are used to train the classifier. Finally, each sample is tested only once.
As a multi-label classification model, we mainly used three measurements to evaluate the predicted results yielded by ten-fold cross-validation, they were accuracy, exact matching, and hamming loss. For formulation, some notations were necessary to define. Given a dataset with n samples and m labels, let Li be a set consisting of true labels of the i-th sample, and Li' be a set consisting of the predicted labels of the i-th sample. The definitions of three measurements were as follows:
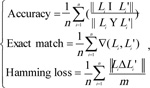 |
(1) |
where ∆ was the symmetric difference operation of Li and Li', and 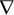 was defined as below:
was defined as below:
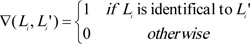 |
(2) |
Clearly, the higher the accuracy and exact match are, the better the performance of the multi-label classification model is, while the lower the hamming loss, the higher the performance.
3. RESULTS AND DISCUSSION
In this study, we proposed a multi-label classifier iMPT-FRAKEL to predict which metabolic pathway type a given compound can participate in. The construction and assessment procedures are illustrated in Fig. (1). In this section, we mainly introduced the evaluation results of iMPT-FRAKEL and compared it with other models to indicate its utility.
3.1. Performance of iMPT-FRAKEL
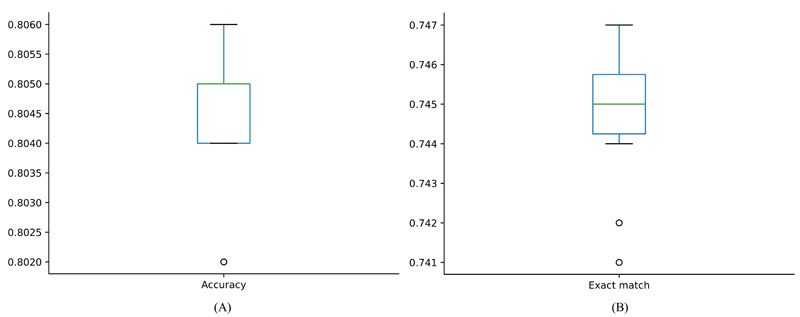
The iMPT-FRAKEL adopted RAKEL and SVM. To build the model with the best performance, we tried several parameter combinations. For example, the main parameter k of RAKEL was set to various values between 2 and 12; another parameter m was set to 10. For SVM, regularization parameter C was set to 0.5, 1 and 2; two kernels: polynomial kernel and RBF kernel were tried, where exponent parameter for polynomial kernel was set to 1, 2 and 3, and the parameter γ for RBF kernel was set to 0.01, 0.02 and 0.03. Models with different parameters were evaluated by ten-fold cross-validation 10 times. Finally, we found that k=12, C=3, RBF kernel with γ=0.03 yielded the best performance. The average of accuracy, exact match and hamming loss are listed in Table 2. They were 0.804, 0.745 and 0.039, respectively. Specifically, the hamming loss values yielded by ten-fold cross-validation ten times were same, they were all 0.039. For accuracy and exact match, their distributions are illustrated in Fig. (2). It can be observed that accuracies were all between 0.802 and 0.806 and exact match values were all between 0.741 and 0.747, indicating that the performance of iMPT-FRAKEL was quite stable for different divisions of the dataset.
| Model | Accuracy | Exact Match | Hamming Loss |
|---|---|---|---|
| RAKEL model (SVM: RBF kernel) (iMPT-FRAKEL) |
0.804 | 0.745 | 0.039 |
| BR model (SVM: RBF kernel) |
0.748 | 0.693 | 0.039 |
| RAKEL model (SVM: polynomial kernel) |
0.787 | 0.716 | 0.046 |
| BR model (SVM: polynomial kernel) |
0.754 | 0.665 | 0.045 |
| RAKEL model (RF) |
0.784 | 0.697 | 0.046 |
| BR model (RF) |
0.706 | 0.648 | 0.044 |
As mentioned above, we also tried another widely used kernel, polynomial kernel, for SVM. The performance of the best model with SVM (polynomial kernel) as the basic classification algorithm is listed in Table 2. The accuracy, exact match and hamming loss were 0.787, 0.716 and 0.046, respectively. Compared with the performance of iMPT-FRAKEL, the accuracy was 1.7% lower, the exact match was 2.9% lower and the hamming loss was 0.7% higher. These results indicated that the selection of RBF kernel as the kernel of SVM was a good choice.
3.2. Comparison of RAKEL Model with Random Forest
The proposed classifier, iMPT-FRAKEL selected SVM as the basic classification algorithm. To elaborate this, selection is proper, we also tried another classic and widely used classification algorithm, RF. For the main parameter, the number of decision trees, we tried various values, including 50, 100, 150 and 200. Models with different parameters were also evaluated by ten-fold cross-validation 10 times. The performance of the best model is listed in Table 2. It can be seen that the accuracy, exact match and hamming loss were 0.784, 0.697 and 0.046, respectively.
Compared with the accuracy and exact match of the iMPT-FRAKEL, the above accuracy and exact match were all lower. As for hamming loss, it was higher than that of iMPT-FRAKEL, indicating that iMPT-FRAKEL was superior to such model. In addition, such model was also inferior to the model with SVM (polynomial kernel) as the basic classification algorithm. As illustrated in Fig. (1), the proposed model (RAKEL model 1 in Fig. (1) was the best RAKEL model for prediction of pathway types of compounds, followed by the RAKEL model with SVM (polynomial kernel) (RAKEL model 2 in Fig. (1) and RAKEL with RF (RAKEL model 3 in Fig. (1). All these implied that the choice of SVM (RBF kernel) was the best choice in a sense for constructing the RAKEL model.
3.3. Comparison of Models with Binary Relevance
Binary Relevance (BR) method [29] is another classic scheme for tackling multi-label classification problems, which builds a binary classification model for each label independently with the one-against-all strategy. We built several multi-label classification models with BR and compared them with RAKEL models. For convenience, these models were called BR models.
The BR model also needs a basic classification algorithm. Likewise, we also employed SVM and RF, as mentioned above. Their same parameter settings mentioned in Section 3.1 and 3.2 were all tried. Each model was assessed by ten-fold cross-validation 10 times. The performance of best BR models with SVM (RBF kernel), SVM (polynomial kernel) and RF is listed in Table 2. It can be observed that with the same basic classification algorithm, RAKEL model always yielded higher accuracy and exact match, about 5% higher, while the hamming loss values of two models were almost at the same level. As illustrated in Fig. (1), RAKEL model had a stronger ability for the prediction of metabolic pathway types of compounds than BR model. All these indicated that the RAKEL algorithm was a good choice for tackling the problem addressed in this study. Furthermore, for BR models, SVM still gave higher performance than RF, which conformed to the results of RAEKL models, further confirming that SVM was the optimal choice for constructing the model.
3.4. User Guide of iMPT-FRAKEL
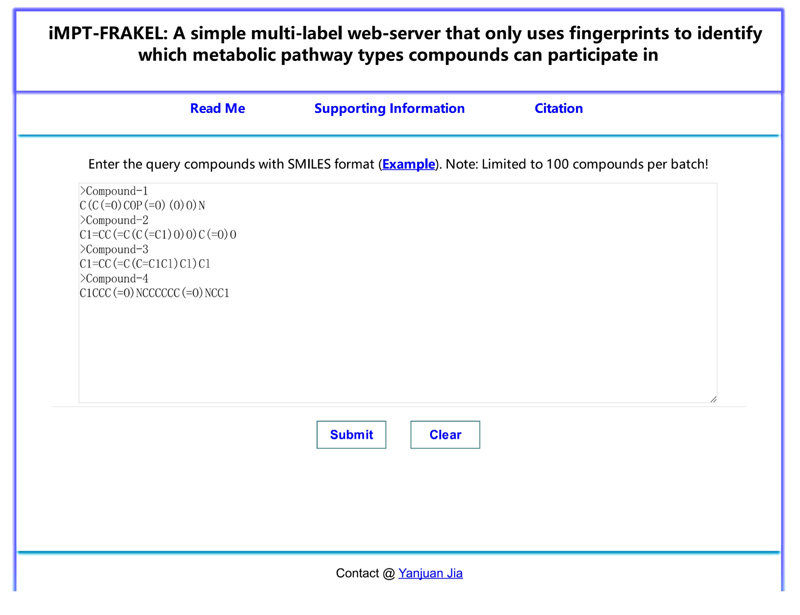
For wide applications of the proposed multi-label classifier, iMPT-FRAKEL, we built its web-server version with the same name. Users can access the web-server iMPT-FRAKEL at http://cie.shmtu.edu.cn/impt/index. Its home page is illustrated in Fig. (3).
In the home page, there are three tabs, say “Read Me”, “Supporting Information” and “Citation”. By clicking “Read Me”, users can obtain the basic information of such web-server, including used methods and parameter settings. Supporting information, such as metabolic pathway types and fingerprints of 4,739 compounds, can be retrieved in the “Supporting Information” tab. The last tab “Citation” lists the reference of such web-server.
To use our web-server for prediction, users should follow the following steps.
1 Use SMILES format to represent each input compound, examples can be found by clicking “Example” button above the input box.
2 Copy the query compounds with SMILES format into the input box and click “Submit” button to submit the query compounds. It is necessary to point out that no more than 100 compounds are permitted each time due to our limited computational power. If users copy wrong information into the input box, click “Clear” button to quickly clear the input box.
3 After a few seconds, users can obtain the predicted results on a new page. Results are divided into two parts. In PART I, predicted metabolic pathway types (represented by tags in Table 1, their detailed names can be found in the top of this page) of each valid compound are listed. Users can download the predicted results by clicking “Result export” button. In PART II, input compounds without fingerprints information are listed. The “Test again” can guide users for another input.
CONCLUSION
This study proposed a simple multi-label classifier to predict the metabolic pathway types of compounds and further built a web-server. Some machine learning algorithms were used to build the classifier, such as RAKEL algorithm and SVM. The experimental results showed that the classifier was quite effective. Compared with the previous classifiers, it was a pure multi-label classifier and had wider applications because it only required the SMILES format of compounds. It is hoped that such classifier can be a useful tool for finding new participants of existing metabolic pathways.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
This study was supported by Natural Science Foundation of Shanghai (17ZR1412500), Science and Technology Commission of Shanghai Municipality (STCSM) (18dz2 271000).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

