
Identification of Collective Expression Pattern of miRNA in the Regulation of Diabetic Retinopathy
Authors Info & Affiliations
Abstract
Background
Diabetic Retinopathy (DR) is a global health problem that is statistically more prevalent in the working age group. MicroRNAs (miRNAs) have been considered downstream molecules involved in several diseases. Recent research has reported that miRs can modulate multiple steps of the pathogenesis of DR.
Aim
This study aimed at identifying significant pathways and genes that cause the dysregulation of the identified miRs to enhance and/or cause development in an individual.
Methodology
Extensive data mining led to the discovery of these novel miRNAs. Functional Enrichment Analysis of these targets was carried out using the MIENTURNET tool through KEGG and WikiPathways databases. The miRNet online tool was used to construct a network analysis.
Results
Nine miRs were found to be associated with DR. We identified four significant pathways, including their involvement in DR: the TGF-beta signaling pathway, insulin signaling pathway, VEGF signaling pathway, and type II diabetes mellitus pathway.
Conclusion
Identifying the targets for DR has the potential for developing new therapies for this chronic disease. The findings of this current work reveal different aspects of the results. Thus, identified miRNAs are found as potential novel biomarkers in identifying the mechanistic approach against DR.
1. INTRODUCTION
MicroRNAs (miRNAs), the name of which comes from “micro” and “repressor,” are non-coding RNA molecules of 19 to 25 nucleotides long that regulate about 60-65% of protein-coding genes in humans [1, 2]. They follow cell type-specified expression patterns and form base pairs with their target messenger RNAs to perform silencing. Evidence proves that the abnormal regulations and alterations in the machinery of miRNA biogenesis are involved in many neovascular ocular disorders. In the nucleus of each cell, RNA Polymerase II transcribes miRNA genes into pri-miRNAs. Drosha, RNase III endonuclease, and DiGeoge Syndrome Critical Region 8 (DGCR8) process the pri-miRNAs into pre-miRNAs. Exportin-5 can bind these pre-miRNAs to the cell cytoplasm. Dicer, RNase III endonuclease, and TAR RNA Binding Protein (TRBP) cleave the pre-miRNA by discarding the closing loop and creating a miRNA duplex. Then, the argonaute family protein (AGO) selects the fully developed miRNA strand and fabricates it in an RNA-Induced Silencing Complex (RISC). Furthermore, miRNAs have the ability to regulate many protein-coding genes. Bioinformatics predictions successfully suggest that miRNAs regulate nearly 30-40% of protein-coding genes. Therefore, the standard production of miRNAs in every cell of the human body is essential [3]. Thus, miRNAs are vital in directing different life processes, including apoptosis, proliferation, differentiation, etc [4]. Recent researchers have discovered the association of abnormal expression of miRs in many chronic diseases, including Diabetic Retinopathy (DR) [5]. Thus, considering their involvement in angiogenesis and neurological and cardio- vascular diseases, miRNAs are seen as budding restorative targets in making effective drugs for future use and as novel biomarkers [6].
Intense scrutinization has resulted in plenty of miRNAs, whose dysregulation is a causative factor associated with ocular disorders. At the same time, a few among them are recognized as in charge of angiogenesis in the retina (e.g., miR-16) [7]. An ocular disease, DR, is a complication of Type 2 Diabetes (T2D) that occurs when the blood vessels in the retina leak blood into the vitreous cavity and result in leakage of blood into the retina [8-10]. The most severe form of diabetes refers to the people who have been suffering from it for more than 15 years. Complete blindness is caused when blood from the iritis travels down to the retina, which causes degeneration. Patients suffering from it have high oxidative stress, superoxide levels, and impairment in the defense system of antioxidants of the retina [11]. Over 462 million individuals suffer from T2D across the globe, which, in turn, makes DR quite a common ocular disease and an essential topic for researchers [12].
The International Diabetes Federation (IDF) assessed the global population with diabetes mellitus (DM) to be 463 million in 2019 and expected it to be 700 million by 2045 [13]. As the most prevalent and particular consequence of DM, DR is one of the primary causes of blindness in the adult working population. According to the Global Burden of Disease Study, DR is the sixth largest cause of blindness and moderate to severe vision impairment in individuals aged 50 and above. From 1990 to 2020, the age-standardized global prevalence of blindness caused by diabetic eye disease grew by 14.9% to 18.5% [14]. The IDF also stated that the global prevalence of DR in 2019 was greater than 25% [15]. Moreover, in 2013, it was reported that the number of adults with diabetes in Africa will increase by 98%, from 12.1 million in 2010 to 239 million in 2030 [16]. Another critical pooled study of different hospital-based research revealed the overall incidence of DR in African countries to be 31.6% and 19.48% in Ethiopia [17, 18].
A prior meta-analysis on the world's overall incidence of DR was carried out over a decade ago, utilizing data from 35 population-based research up to 2008. There is a need for current data, as some significant changes in the epidemiologic aspects of DR have surfaced in recent years. Firstly, a decline in DR prevalence was suggested, particularly in wealthy countries. This was most likely due to enhanced awareness and better systemic control in diabetic individuals. Secondly, most of the research included in the recent meta-analysis was from European ancestry populations [19]. Since 2008, there has been significant growth in population-based research in other regions, notably Asia, which represents roughly half of the worldwide DM population. China (116 million) and India (77 million) have the biggest number of individuals with diabetes, reflecting Asia's rapid economic expansion and urbanization over the last decade, as well as significant lifestyle and nutritional changes. The worldwide DR burden is predicted to increase in 2045, hitting nations disproportionately in the Middle East, North Africa, and the Western Pacific. This makes it necessary for humankind to be well aware of the statistical and geographical spread of DR for better prevention and to develop healthcare measures [20].
The current study uses bioinformatics tools to determine the miRs associated with the DR. Various studies have reported a series of genes, like VEGF, erythropoietin, cytokines, etc., being the target of the miRs. In this study, the selected miRs putative targets, functional enrichment analysis, and pathway enrichment are retrieved. Therefore, the recognition of the miRs involved in DR by using bioinformatics tools and putative target retrieval is inevitable for their network construction and pathway enrichment. Pathway enrichment would clarify and help us detect the role played by that miR in the disease. It also provides us with the list of genes involved in a particular pathway correlated with DR. To make a substantial prediction, two different tools, miRTarBase and TargetScan, are used to perform all the mentioned investigations and analyses.
2. MATERIALS AND METHODS
2.1. miRNA Identification and Retrieval of their Putative Targets
The involvement of several miRs in the development of ocular disorders was reported. After extensive data mining, as depicted in Fig. (1), 9 miRs were found to be associated with the DR. miRBase database was used to find the sequence and annotation of these miRs. This database is a searchable database of published miRNA sequences and annotations. All the entries in the miRBase sequence database have a predicted hairpin miRNA portion transcript with information and sequence of the mature miR; both are available for search and browsing. Two databases, miRTarBase and TargetScan, were used to find the targeted genes for every selected miR. miRTarBase is one of the substantially accessible handbook-curated target databases containing more than 65,000 interactions of miRNA-gene. Targets extracted from specified and high throughput investigations like microarrays and proteomics are incorporated. It provides data obtained from 3 CLIP-Seq and 12 Degradome-Seq studies, more than any other accessible databases. TargetScan anticipates the biological targets of miRNAs by probing for the existence of the conserved 8mer, 7mer, and 6mer sites that complement the seed region of each miRNA. The latest version uses an upgraded procedure to anticipate targeting efficacy and a 3' UTR profile stipulating the fraction of mRNA accommodates each site and utilizes upgraded miRNA families. Web tool MIENTURNET can also be utilized for the same purpose; it provides us with the collective targeted genes from miRTarBase [21] and TargetScan [22], respectively, for all the miRs.
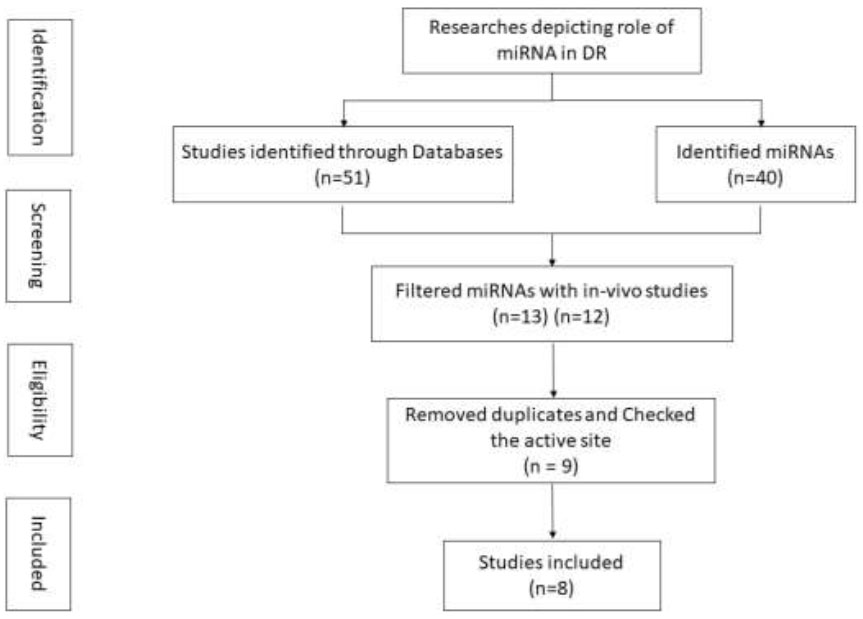
Flowchart depicting study selection.
| miR | Dysregulation | miRBase Accession | miR Mature Sequence | Author, Year |
|---|---|---|---|---|
| hsa-miR-126-3p | Downregulated | MIMAT0000445 | UCGUACCGUGAGUAAUAAUGCG | (Beatrix, 2011) |
| hsa-miR-145-5p | Downregulated | MIMAT0000437 | GUCCAGUUUUCCCAGGAAUCCCU | (Jingjing, 2019) |
| hsa-miR-146a-5p | Upregulated | MIMAT0000449 | UGAGAACUGAAUUCCAUGGGUU | (Eun-Ah, 2016) |
| hsa-miR-150-5p | Downregulated | MIMAT0000451 | UCUCCCAACCCUUGUACCAGUG | (Liu, 2015) |
| hsa-miR-155-5p | Upregulated | MIMAT0000646 | UUAAUGCUAAUCGUGAUAGGGGUU | (You, 2018) |
| hsa-miR-200b-3p | Upregulated | MIMAT0000318 | UAAUACUGCCUGGUAAUGAUGA | (Metin, 2018) |
| hsa-miR-21-3p | Upregulated | MIMAT0004494 | CAACACCAGUCGAUGGGCUGU | (Mazzeo, 2019) |
| hsa-miR-221-3p | Upregulated | MIMAT0000278 | AGCUACAUUGUCUGCUGGGUUUC | (Wang, 2020) |
| hsa-miR-27b-3p | Downregulated | MIMAT0000419 | UUCACAGUGGCUAAGUUCUGC | (Metin, 2018) |
2.2. Enrichment Analysis
Gene Enrichment Analysis is a technique of recog- nizing the pathways associated with the identified genes predicted to be associated with a particular disorder or disease. MIENTURNET [23], an in-silico tool, was utilized for functional enrichment analysis of miRNA-putative targeted genes. This tool yielded separate analyses of functional enrichment results from two different approaches of the Kyoto Encyclopedia of Genes and Genomes (KEGG) [24] and WikiPathways [25]. Kyoto Encyclopedia of Genes and Genomes database precisely dispenses high-scale molecular datasets created by genome sequencing and high-throughput experimental technologies. It is broadly used as a reference database and knowledge base for integrating and elucidating large-scale datasets. It is being discussed in relation to more practical implementations that combine diseases in humans, drugs, and other interconnected things. WikiPathways is a conventional graphical pathway modifying tool built on MediaWiki software. It is amalgamated with different databases and contains all the essential genes, proteins, and small molecule systems. It is an open, synergistic platform that is dedicated to the curation of biological pathways, thereby attracting a broader engagement from the research society.
2.3. Network Construction
For the network construction of miRNAs-target genes, miRNet [26] was used. A miRNA-centric network visual analytics is a platform providing support for various inputs and statistics. It accepts a catalog of miRNAs, miRNA-SNPs, genes, transcription factors, small molecules, ncRNAs, diseases, epigenetic modifiers, any combinations, or a data table from microarray, RNAseq, or RT-qPCR investigations. It has comprehensive functional anno- tations, amalgamating data from 14 discrete miRNA databases. It is mainly used for designing miRNA-target interaction systems and forming high-performance network visual analytics. miRNet selected nine miRNA (hsa-miR-126-3p, hsa-miR-145-5p, hsa-miR-146a-5p, hsa-miR-150-5p, hsa-miR-155-5p, hsa-miR-200b-3p, hsa-miR-21-3p, hsa-miR-221-3p, and hsa-miR-27b-3p) and their 10735 targeted genes with a degree filter of 1.0 to construct a network of 18973 edges. Here, the pathway enrichment analysis of four critical pathways was performed to find the genes that were particularly present in their pathways.
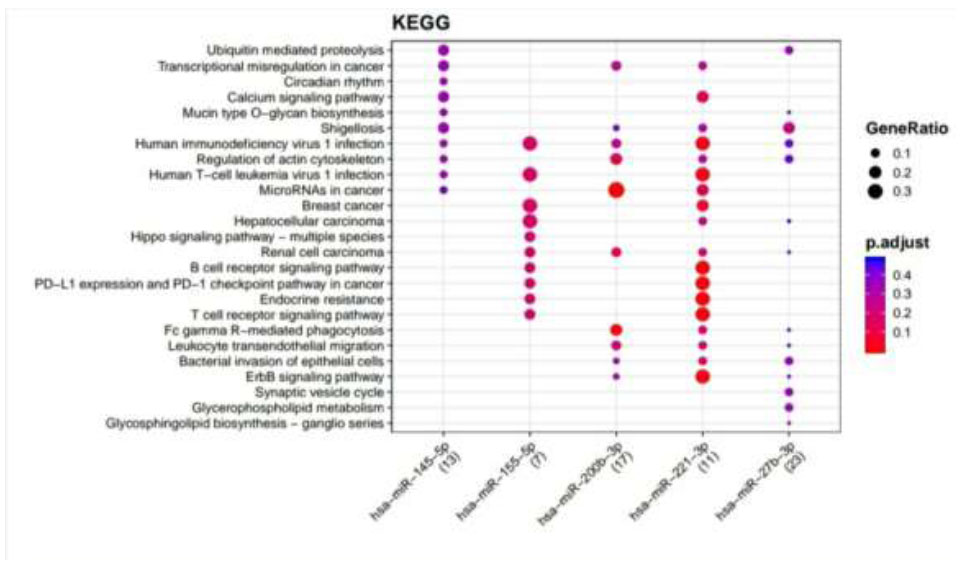
KEGG pathways enriched for DR putative miRNA target genes of TargetScan. Functional Enrichment Analysis of target genes for hsa-miR-145-5p, hsa-miR-155-5p, hsa-miR-200b-3p, hsa-miR-221-3p, and hsa-miR-27b-3p were retrieved from TargetScan by KEGG. The color change and size of the circles depict the adjusted p-value (reflects the degree of certainty with which the pathway is enriched) and gene ratio (a percentage that consists of all Differentially Expressed Genes in the Gene Ontology), respectively.
3. RESULTS
3.1. Identification of Involved miRs and Retrieval of Putative Targets
An extensive literature review and survey led to the screening of nine miRs whose association with DR had been reported (Fig. 1). Details, such as miR name, accession ID, miR mature sequence, etc., were retrieved through the miRbase database (Table 1). miR putative targeted genes were retrieved from miRTarBase and TargetScan databases through MIENTURNET, an online tool. It provided information on the targeted genes for the identified miRs after the statistical analysis. Considering the default threshold value, miRTarBase and TargetScan databases yielded 349 and 1071 genes, respectively, for all nine miRs collectively.
3.2. Functional Enrichment Analysis of Putative Target Genes
Retrieved putative genes from miRTarBase and TargetScan were further subjected to Functional Enrichment Analysis by two different databases, KEGG and Wikipathways, through MIENTURNET. Functional Enrichment Analysis by KEGG for TargetScan putative target genes provided us with more than 50 pathways, amongst which the top 25 pathways are displayed in Fig. (2) while taking into consideration the default threshold value. The size of the circle in the graph represents genes being involved in the pathway. The larger the size, the greater the number of genes involved in the pathway, and vice versa. Amidst this ubiquitin-mediated proteolysis pathway, circadian rhythm, mucin-type O-glycan biosynthesis, regulation of actin cytoskeleton, B cell receptor signaling pathway, endocrine resistance, and ErbB signaling pathway associated with DR were enriched more in hsa-miR-221-3p. The Functional Enrichment Analysis for miRTarBase putative target genes yielded more than 80 pathways, amongst which the top 29 pathways are plotted in Fig. (3) while taking into consideration the default threshold value. Among them, the chemokine signaling pathway, neurotrophin signaling pathway, FOXO signaling pathway, EGFR tyrosine kinase inhibitor resistance, AGE−RAGE signaling pathway in diabetic complications, cellular senescence, endocrine resistance, relaxin signaling pathway, cysteine and methionine metabolism, and biosynthesis of amino acids, which are associated with DR, were exceedingly enriched in hsa-miR-126-3p, hsa-miR-146a-5p, hsa-miR-155-5p, hsa-miR-221-3p, hsa-miR-145-5p, hsa-miR-200b-3p, and hsa-miR-27-3p.
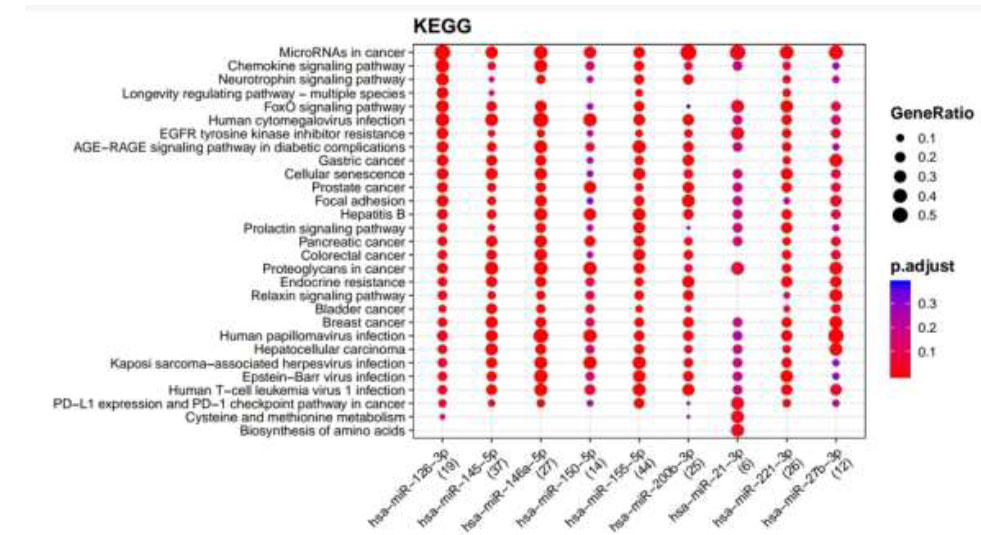
KEGG pathways enriched for DR putative miRNA target genes of MiRTarBase. Functional Enrichment Analysis of target genes for hsa-miR-145-5p, hsa-miR-155-5p, hsa-miR-126-3p, hsa-miR-146a-5p, hsa-miR-150-5p, hsa-miR-21-3p, hsa-miR-200b-3p, hsa-miR-221-3p, and hsa-miR-27b-3p retrieved from MiRTarBase by KEGG. The color change and size of the circles depict the adjusted p-value (reflects the degree of certainty with which the pathway is enriched) and gene ratio (a percentage that consists of all Differentially Expressed Genes in the Gene Ontology).
Functional Enrichment Analysis by WikiPathways for TargetScan putative target genes rendered more than 50 pathways, amongst which the top 18 pathways are presented in Fig. (4) while taking into consideration the default threshold value. The size of the circle in the graph represents genes being involved in the pathway. The larger the size, the more genes are involved in that pathway, and vice versa. Among them, the EGF/EGFR signaling pathway, TNF alpha signaling pathway, insulin signaling, GPR40 pathway, VEGFA−VEGFR2 signaling pathway, endoderm differentiation, ErbB signaling pathway, Wnt signaling pathway, Oncostatin M signaling pathway, IL−7 signaling pathway, which are associated with DR, were considerably enriched more in hsa-miR-221-3p and hsa-miR-155-5p. Functional Enrichment Analysis for miRTarBase putative target genes furnished more than 80 pathways, amongst which the top 29 pathways are highlighted in Fig. (5) while taking into consideration the default threshold value. Among them, IL−5 signaling pathway, chemokine signaling pathway, DNA damage response (only ATM dependent), Oncostatin M signaling pathway, focal adhesion, VEGFA−VEGFR2 signaling pathway, AGE/RAGE pathway, B Cell receptor signaling pathway, TGF−beta signaling pathway, apoptosis modulation, and signaling, methylation pathways, and methionine De Novo and salvage pathway, which are associated with DR, were found to be enriched in hsa-miR-126-3p, hsa-miR-145-5p, hsa-miR-146a-5p, hsa-miR-150-5p, hsa-miR-155-5p, hsa-miR-200b-3p, and hsa-miR-221-3p.
Two different databases, KEGG and WikiPathways, were utilized for Functional Enrichment analysis. Both databases work on different methodologies and algorithms to exploit the results. The overlapped study of the results assembled from the two allowed the retrieval of common pathways that were found to be vital in the prognosis of DR.
A Venn diagram is drawn to retrieve the number of common pathways found for the target genes of miRTarBase and TargetScan in KEGG and WikiPathways. The Venn diagram (Fig. 6) shows that out of 218 and 176 pathways from miRTarBase and TargetScan, respectively, 158 pathways were found to be common through the KEGG database. Out of these 158 overlapped pathways, DR-associated pathways like the TGF-Beta signaling pathway, insulin signaling pathway, VEGF signaling pathway, Type II diabetes mellitus, glucagon signaling pathway, insulin resistance, diabetic cardiomyopathy, and insulin secretion were the similar specific pathways in both the databases. The Venn diagram (Fig. 7) illustrates that out of 289 and 128 pathways from miRTarBase and TargetScan, respectively, 124 were found common through the WikiPathways database. Of these 124 overlapped pathways, DR-associated pathways like insulin signaling, VEGFA-VEGFR2 signaling pathway, and TGF-beta pathway were similar and specific in both databases.
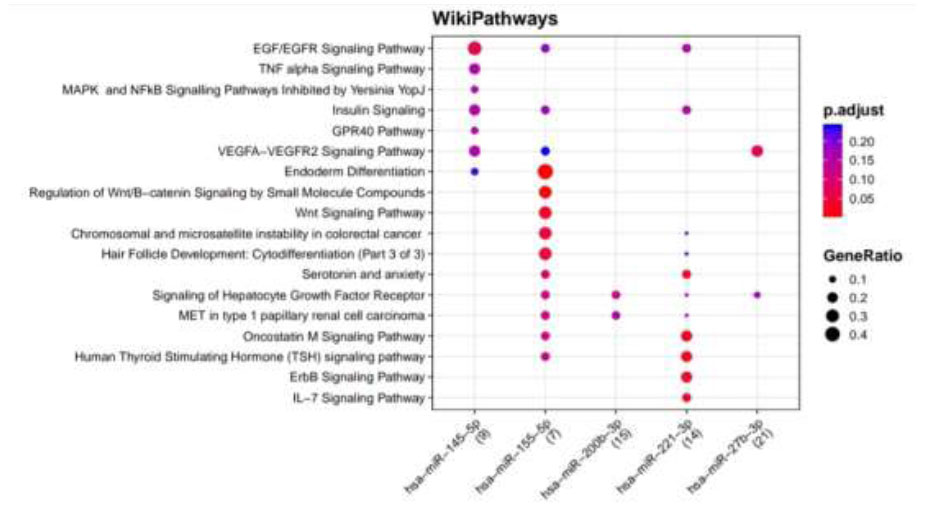
WikiPathways identified pathways enriched for DR putative miRNA target genes of TargetScan. Functional Enrichment Analysis of target genes for hsa-miR-145-5p, hsa-miR-155-5p, hsa-miR-200b-3p, hsa-miR-221-3p, and hsa-miR-27b-3p retrieved from TargetScan by WikiPathways. The color change and size of the circles depict the adjusted p-value (reflects the degree of certainty with which the pathway is enriched) and gene ratio (a percentage that consists of all Differentially Expressed Genes in the Gene Ontology).
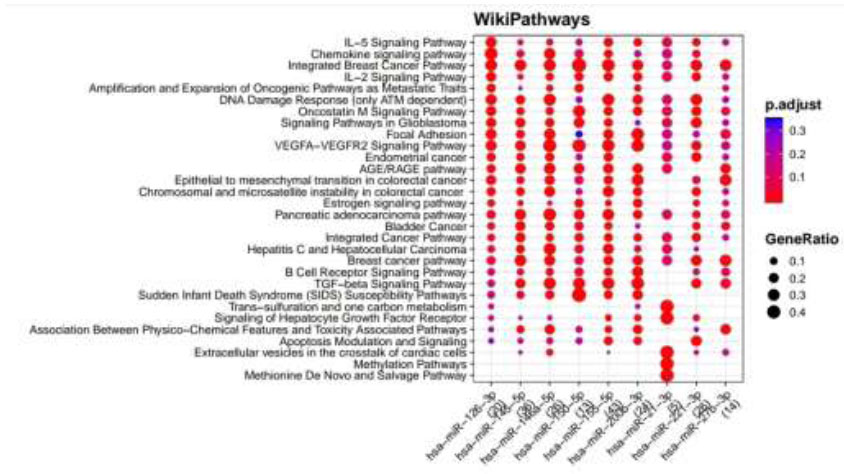
WikiPathways identified pathways enriched for DR putative miRNA target genes of MiRTarBase. Functional Enrichment Analysis of target genes for hsa-miR-145-5p, hsa-miR-155-5p, hsa-miR-126-3p, hsa-miR-146a-5p, hsa-miR-150-5p, hsa-miR-21-3p, hsa-miR-200b-3p, hsa-miR-221-3p and hsa-miR-27b-3p retrieved from MiRTarBase by WikiPathways. The color change and size of the circles depict the adjusted p-value (reflects the degree of certainty with which the pathway is enriched) and gene ratio (a percentage that consists of all Differentially Expressed Genes in the Gene Ontology).
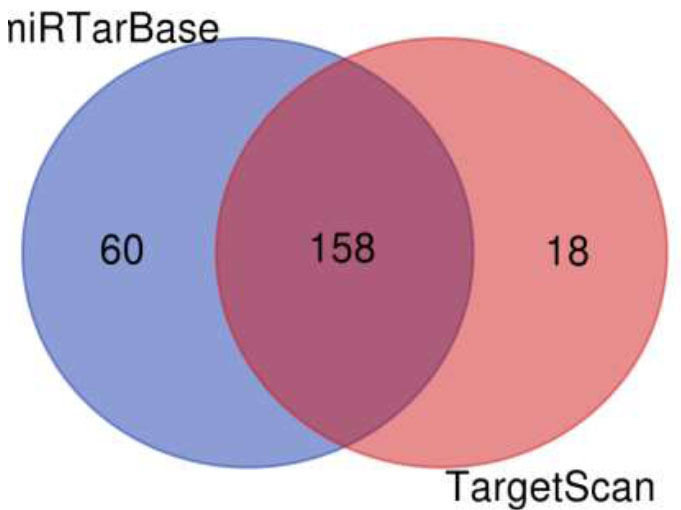
Venn diagram of pathways by KEGG for putative targets.
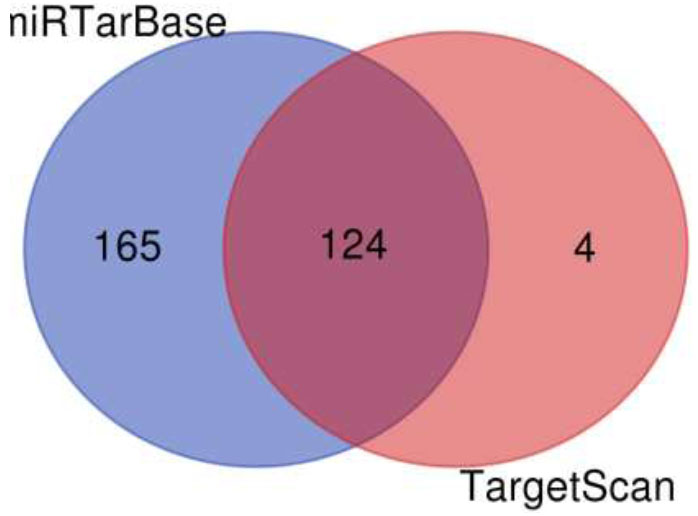
Venn diagram of pathways by wikipathways for putative targets.
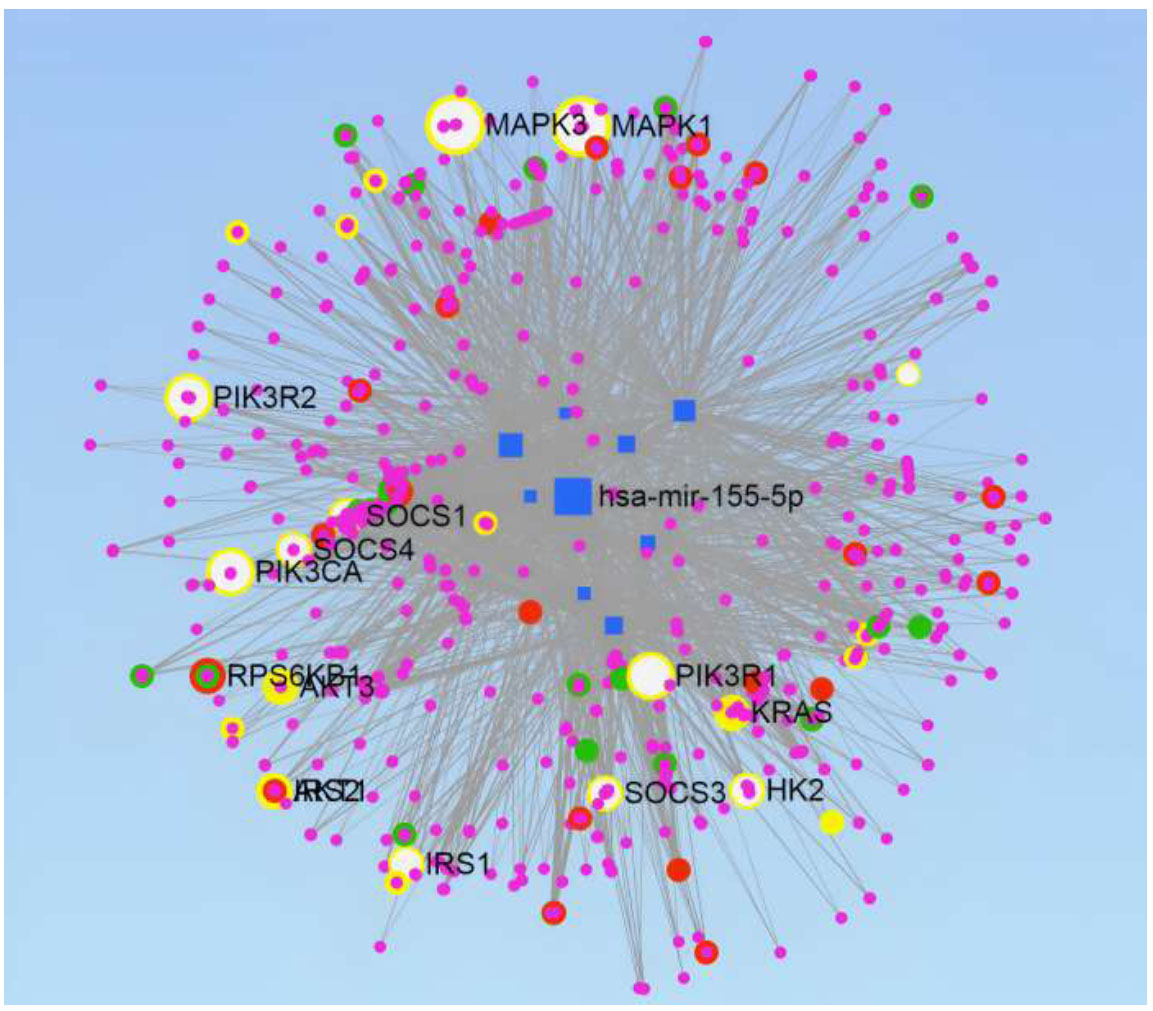
miRNAs-target genes network construction. Genes involved in the four pathways are highlighted:- TGF-beta signaling Pathway-Green, Insulin Signaling Pathway-Red, VEGF Signaling Pathway-Yellow, and Type II Diabetes Mellitus pathway-White.
3.3. miR-Gene Network Construction and Analysis
miRNet tool is used to construct the network of miRNAs-target genes. It illustrated the network analysis of the selected miRs, which targeted 10735 genes, produced 18973 edges, and provided us with the various pathways involved in DR. Degree Filter of 2.8 was used to construct the final network, which consisted of 2068 genes and 7351 edges (Fig. 8). miRNet also supports pathway enrichment from different databases. The KEGG database was used with a hypergeometric sampling algorithm for target gene enrichment; more than 60 pathways were enriched with a False Discovery Rate (FDR) < 20%. The FDR quantifies the fraction of incorrect findings amongst the set of significant hypothesis tests. Adjusted p-values are commonly used to estimate this amount. Amongst these pathways, four significant pathways were identified, which are already known for their involvement in DR and have been reported in the literature. The genes intricated in the TGF-beta signaling pathway, insulin signaling pathway, VEGF signaling pathway, and type II diabetes mellitus pathway are highlighted in the network constructed by green, red, yellow, and white color, respectively (Fig. 8).
4. DISCUSSION
DR is a congenital ocular disease that develops in about one-third of diabetic patients and causes vision loss in the middle-aged population worldwide [27]. Early diagnosis and treatment of this condition can significantly improve the quality of life for patients by decreasing complications, such as blindness and cataracts. A major challenge in treating DR is its slow development, which often leads to delayed diagnosis. Thus, it is now being acknowledged as a global epidemic [28]. Early symptoms of DR comprise microaneurysms, exudates, hemorrhages, and angiogenesis. Intense data mining led to the recognition of nine important miRNAs whose role was verified in the prognosis of DR. Putative target genes retrieved through miRTarBase and TargetScan databases yielded about 349 and 1071 genes, respectively, for hsa-miR-126-3p, hsa-miR-145-5p, hsa-miR-146a-5p, hsa-miR-150-5p, hsa-miR-155-5p, hsa-miR-200b-3p, hsa-miR-21-3p, hsa-miR-221-3p, and hsa-miR-27b-3p miRs collectively.
A survey done in 2021 stated that the prevalence of DR is 16.9% in India [29]. To combat the implications of DR, it is mandatory to identify its pathogenesis. Oxidative stress and alterations in cellular metabolism are caused by prolonged T2D, which further leads to retinal degeneration [30]. In the course of time, this stimulates the expression of hypoxia-induced growth factors like VEGF, which advances retinal neovascularization (NV). Retinal NV encourages hemorrhage; the ultimate outcome is complete vision loss [31]. Functional Enrichment Analysis of the retrieved putative genes produces two separate datasets of enriched pathways by KEGG and WikiPathways. These findings highlighted the develop- mental role of the TGF-beta signaling pathway, insulin signaling pathway, VEGF signaling pathway, and type II diabetes mellitus pathways (FDR < 20%) in DR. Thus, the genes involved in these pathways are suspected to be the prime functioning unit of the disease. The results are also supported by studies where numerous changes in miRNA expression or function are observed as essential for the development of insulin resistance in Type 2 Diabetes (T2D) in a variety of insulin-targeting organs. MiRNAs have been perceived indirectly as modulators of inflammation-induced insulin resistance by controlling/tuning the activity of innate immune cells in insulin-target organs [32]. Multiple studies indicate that in diabetes, retinal vasculature becomes dependent on a slight increase in TGF-signaling via activin receptor-like kinase 5 for maintaining early integrity. Increased TGF-signaling may protect against fast retinopathy progression and should not be the target of inhibitory therapies [33].
A review carried out in 2013 provided detailed information on VEGF and its function in the etiology of DR. Laser photocoagulation is stated to be the most preferred therapy for proliferative DR. However, it is still found to be ineffective in many individuals. Alternatively, anti-VEGF medicines are slowly gaining popularity among retina experts. Although only a few of these medications have FDA approval for intraocular use, like ranibizumab and pegaptanib, several others are currently utilized off-label in clinical settings, like bevacizumab. These anti-VEGF medications are now utilized as an adjunct therapy to laser treatment, but they are increasingly being employed as a primary treatment [34]. Due to the fact that miRNAs play a vital part in the etiology of type 2 diabetes, delivery of miRNA mimics or antimiRs in-vivo has recently resulted in favorable regulation of glucose and lipid metabolism.
Furthermore, various cell culture-based therapies have revealed that miRNAs have beta-cell regeneration capability. Despite this, only a handful of miRNA-based treatment methods have reached the clinical stage. As a result, the contributions of future studies would help in discovering the miRNA therapies in the treatment of T2DM [35].
Varied investigations have proved the importance of miRs in the diagnosis of severe disorders. Depending upon the type of disorder to be examined, serum or blood instances from affected and unaffected ones are subjected to analysis, and the selected miRs are observed for either up-regulation or down-regulation. A study titled “Hypoxia-Induced Deregulation of miR-126 and its Regulative Effect on VEGF and MMP-9 Expression” was carried out, which manifested the downregulation of miR-126 in the serum of TYPE1 and TYPE2 diabetes mellitus patients by targeting SPRED-1, PIK3R2, and VECAM-1 gene [36]. Similarly, there were other related investigations to discover the role of miR-150, miR-155, miR-221, miR-27b; miR-126, miR-146, miR-21; miR-23a, miR-214, and miR-200b in ocular diseases like DR, wet Age-Related Macular Degeneration (AMD) and Retinopathy of Prematurity (ROP), respectively [37].
In recent years, a lot of work has been conducted worldwide to identify the association of miRs with ocular disorders. Particularly, circulating miRs have been identified through histological assessment to take part and play a significant role in the occurrence of DR [38]. This scanning recommends the usage of miRs as novel biomarkers in predicting the stage of DR and diabetes mellitus [39]. Online tools of bioinformatics are a recent development that provides us with an in-silico approach that combines bio-physiology with a computational outlook. This development assists in lessening the burden of vital experimental labs and allows us to perform accurate predictions. miRscan regulates many protein-coding genes. Therefore, the standard production of miRs in every cell of the human body is essential.
CONCLUSION
In today's era, miRNAs are associated with ocular disorders and are seen as potential novel biomarkers in the identification and finding of stages of disorders. This study can help decrypt the budding involvement of miRNAs in the global epidemic, DR. The network construction identified four major pathways, and the pathway enrichment of these pathways identified the genes intricated in them. These genes and pathways together can aid in succeeding investigations in developing potential drugs against DR in the near future. Based on this primitive finding, a hypothesis may be drawn: a group of miRNAs collectively work in the expression of any ocular disorder, and it may be concluded that the entire responsibility can never be laid on the shoulders of a single miRNA. This study may aid in examining if any miRNA associated with diabetes later also assists in the prognosis of DR. Furthermore, studies can be carried out to establish the interconnection of the identified miRs with other ocular disorders.
AUTHORS’ CONTRIBUTION
It is hereby acknowledged that all authors have accepted responsibility for the manuscript's content and consented to its submission. They have meticulously reviewed all results and unanimously approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| DR | = Diabetic Retinopathy |
| miR(s) | = MicroRNA(s) |
| DGCR8 | = DiGeoge Syndrome Critical Region 8 |
| TRBP | = TAR RNA Binding Protein |
| AGO | = Argonaute family protein |
| RISC | = RNA- Induced Silencing Complex |
| T2D | = Type 2 Diabetes |
| T2DM | = Type 2 Diabetes Mellitus |
| DM | = Diabetes Mellitus |
| IDF | = International Diabetes Federation |
| VEGF | = Vascular endothelial growth factor |
| KEGG | = Kyoto Encyclopedia of Genes and Genomes |
| UTR | = Untranslated Region |
| SNP | = Single nucleotide polymorphisms |
| ncRNA | = Non-coding RNAs |
| RT-qPCR | = Quantitative reverse transcription polymerase chain reaction |
| ErbB | = Erythroblastic Oncogene B |
| FOXO | = Forkhead box transcription factors |
| EGFR/EGF | = epidermal growth factor receptor |
| AGE/RAGE | = Receptor for Advanced Glycation End Products |
| TNF | = Tumor Necrosis Factor |
| IL | = Interleukin |
| ATM | = Ataxia Telangiectasia Mutated Gene |
| TGF | = Transforming growth facto |
| FDR | = False Discovery Rate |
| NV | = Neovascularization |
| FDA | = Food and Drug Administration |
| AMD | = Age-Related Macular Degeneration |
| ROP | = Retinopathy of Prematurity |
AVAILABILITY OF DATA AND MATERIALS
The data supporting the article's findings is available in with in the article (Table 1).

