
In Silico Study of Dibenzylidene Cyclohexanone-Based Curcumin Analogs as Potential Inhibitors of Breast Cancer Receptor Proteins
Abstract
Background
Cancer is one of the leading causes of death worldwide, accounting for approximately 10 million deaths annually. The most prevalent type of cancer in women is breast cancer, and there are not any prospective vaccinations available for the treatment of this disease.
Objective
This study aimed to identify potent substances derived from natural products, such as curcumin analogs, which are crucial for enriching drug discovery, particularly in the prevention of breast cancer.
Methods
This study utilized twelve novel curcumin analogs, specifically dibenzylidene-cyclohexanones, to predict their biological activity against breast cancer. Based on Lipinski's rule of five, selected compounds were screened using ADMETlab 3.0 to assess their drug-likeness properties. Then, the selected compounds were subsequently subjected to pharmacophore modeling using LigandScout, followed by molecular docking studies with the human estrogen receptor alpha (ERα; PDB ID: 2IOG) using AutoDock. Curcumin and tamoxifen were included as reference compounds for comparison.
Results
Based on the research conducted, all of the curcumin analogs met the criteria of Lipinski’s rule of five, except compound 12. Compound 4 demonstrated the best potential as an anticancer agent against ERα, with a pharmacophore fit-score of 36.87 based on pharmacophore modeling and binding energy of -11.10 kcal, which was higher than tamoxifen (-10.45 kcal/mol) and curcumin (-9.18 kcal/mol) based on a molecular docking study.
Conclusion
Exploring curcumin analogs as potential anti-breast cancer agents is crucial for drug discovery and development. This study suggests that curcumin analog compound 4 can act as a potent inhibitor against ERα. However, further in vitro studies are required to confirm the efficacy of this compound.
1. INTRODUCTION
Breast cancer, as defined by the World Health Organization (WHO), is a disease characterized by the uncontrolled growth of abnormal breast cells that can form tumors. If left untreated, tumors have the potential to spread throughout the body and become fatal.
Breast cancer cells first multiply in the milk ducts and/or the milk-producing lobules of the breast. In 2022, 2.3 million women were diagnosed with breast cancer, and 670,000 people died from the disease worldwide. Breast cancer can affect women at any age after puberty, with its incidence increasing as women grow older [1].
The degree to which the cancer has progressed to lymph nodes or other bodily areas will depend on its subtype. There are many treatments available for breast cancer. Breast cancer subtypes based on the expression of the hormone receptors are categorized into four categories: human epidermal growth factor receptor-2 positive (HER-2+), progesterone receptor positive (PR+), estrogen receptor-positive (ER+), and those that lack expression of all three receptors, known as triple-negative breast cancer (TNBC), which is more aggressive than other subtypes [1, 2].
Estrogen receptor (ER) and progesterone receptor (PR) are hormones that are essential for regulating key physiological processes, such as sexual maturation (including breast development), pregnancy, postpartum, and menopause. However, normal breast cells can become cancerous when DNA damage occurs, leading to rapid growth and uncontrolled spread, often influenced by related hormones [3]. Alpha (α) and beta (β) estrogen receptors are encoded by two genes, ESR1 and ESR2, respectively [4]. Examples of endocrine (hormone) therapies that are effective in treating malignancies expressing ER and/or PR include tamoxifen and aromatase inhibitors. Chemotherapy is required for “hormone receptor negative” cancers that do not express either ER or PR [1].
Much research has been carried out to find and develop drugs that can inhibit this highly aggressive cancer [5-8]. One of the most challenging obstacles in treating breast cancer, which eventually results in death, is multidrug resistance. Therefore, an in-depth understanding and analysis of the molecular basis of cancer resistance is required, and the development of new effective drugs is urgently needed [9].
Natural materials have been used as a source of medicine for many years. Today, approximately half of all pharmaceutical drugs are still derived from natural sources. These products are usually isolated from plants, marine flora, and microorganisms. Their therapeutic potential can be enhanced through structural modification [10, 11].
Curcumin is a major bioactive compound found in rhizomes and has attracted significant attention from researchers due to its diverse biological activities. It has shown potential benefits in conditions, such as cardiovascular diseases, diabetes mellitus, etc [11, 12]. Previous studies reported that curcumin has the potential to be an anti-breast cancer agent because it affects the number of phenotypes through mechanisms of action, such as (a) P-glycoprotein activity inhibition and drug resistance reduction, (b) cell cycle induction, (c) ferroptosis and apoptosis initiation, and (d) control of the epithelial-mesenchymal transition (EMT) [13, 14].
Other reports demonstrated that curcumin exhibits its anticancer properties via a complex molecular signaling network that includes those related to HER-2, ER, and proliferative pathways. In addition, experimental studies have reported that curcumin regulates genes and microRNAs linked to cell phase and apoptosis in breast cancer cells [15]. However, curcumin is known to have a poor pharmacokinetic profile, and its systemic bioavailability is low due to limited absorption, rapid metabolism, and rapid excretion. One way to slow down the metabolism of curcumin is to explore new curcumin-derived compounds with lower toxicity yet increased effectiveness [16].
Structural modification of curcumin is expected to serve as a next-generation drug candidate for cancer therapy. Several research works have been carried out to explore the effects of curcumin and its analogs as cancer therapy, especially in breast cancer. Several curcumin analogs have been synthesized and tested for their anticancer activity using in-vitro methods in MCF-7 cells, and it was found that some curcumin analogs exhibit toxicity even at small concentrations [8, 17].
A previous study conducted by Shen et al. (2020) reported that curcumin analog with mono-carbonyl (acetone) B14 has antitumor activity and potent selectivity for MCF-7 and MDA-M-231 cells with IC50 values of 8.84 and 8.33 μmol/L, respectively. Its IC50 value for MCF-10A breast epithelial cells was 34.96 μmol/L. Moreover, B14 demonstrated better bioavailability compared to curcumin [18]. Furthermore, monocarbonyl curcumin analogues have been shown to have enhanced intestinal permeability and water solubility, as well as good chemical and metabolic stability linked to improved bioavailability [19].
Based on previous studies and the global prevalence of breast cancer, curcumin analogs, particularly monocarbonyl curcumin analogs, show promising potential as anti-breast cancer agents. The findings of this study can be a source of updated information for researchers on the topic of curcumin analogs as potential breast cancer agents. In a previous study, the curcumin analogs with di-benzylidene-cyclohexanones (Fig. 1) [17, 18] were tested for their antioxidant activity. In this study, we aim to evaluate the pharmacokinetic and ADMET profiles of these compounds through virtual screening, as well as assess their interaction with human estrogen receptor alpha (ERα), a key breast cancer receptor, using pharmacophore modeling and molecular docking in an in silico approach. This study is preliminary research, which is expected to continue with synthesized and laboratory tests.
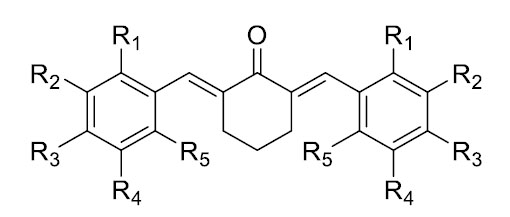
Structure template for dibenzylidene-cyclohexanones.
2. MATERIALS AND METHODS
2.1. Analysis of Drug-Likeness Properties
The evaluation of drug-likeness properties was performed based on Lipinski’s rule of five (molecular weight (MW) ≤ 500 Daltons, logP ≤ 5, hydrogen bond acceptors (HBA) ≤ 10, and hydrogen bond donors (HBD) ≤ 5) [20] using ADMETlab 3.0 (https:// admetlab3. scbdd.com/) [21]. Table 1 presents the tested ligands with their structures shown in Fig. (1) as a template. This study used canonical SMILES generated by MarvinSketch after drawing the structures of the compounds.
2.2. Pharmacophore Modeling
The pharmacophore model was carried out using LigandScout v4.4.1 (licensed by Padjajaran University). LigandScout employs the Espresso algorithm, which involves multiple stages, such as clustering and conformation generation. The Ligandscout algorithm creates a pharmacophore on each training molecule and then aligns the test molecule using this pharmacophore [22]. Therefore, pharmacophore modeling started with model validation, which is crucial for virtual screening as it helps predict the possible pharmacophore activities at the receptor. In this process, a 1:4 ratio of active compounds to decoy compounds obtained from the DUD-E database (https://dude.docking.org/) was used. This process ensures the selection of models with sufficient accuracy [23].
The pharmacophore validation process yielded a model based on the structure of the active compound, which was subsequently used to screen curcumin analog test compounds. The model was based on the receiver operating characteristic (ROC) curve, true positive (TP), and false positive (FP). The ROC curve illustrates the model's ability to distinguish between active and inactive substances, and this performance is measured by the area under the ROC curve (AUC) [22]. An AUC value close to 0 indicates a poor classifier, as the model incorrectly ranks decoys (compounds that should be inactive) higher than active compounds. Conversely, an AUC value close to 1 indicates an excellent classifier, correctly prioritizing active compounds over decoys. In addition, other statistical parameters like goodness-of-hit score (GH), enrichment factor (EF), and accuracy (ACC) will determine the performance of the model [23, 24].
The 3D structures of these compounds were constructed using the ChemDraw application. They were then combined using Discovery Studio and saved in .sdf format. The hit compounds were selected based on their pharmacophore fit score values.
2.3. Molecular Docking
2.3.1. Selection and Retrieval of Receptors
Literature was searched for possible targets for anti-cancer treatments. The three-dimensional structure (in PDB format) of each of the selected receptors was retrieved from the RCSB protein data bank (PDB) (https://www.rcsb.org/) using PBD ID 2IOG (human estrogen receptor alpha ligand-binding domain in complex with compound 11F) [25].
| Compounds | R1 | R2 | R3 | R4 | R5 |
|---|---|---|---|---|---|
| Compound 1 | -H | -Br | -OCH3 | -H | -H |
| Compound 2 | -COOH | -H | -H | -H | -H |
| Compound 3 | -Cl | -H | -H | -H | -F |
| Compound 4 | -Cl | -H | -Cl | -H | -H |
| Compound 5 | -OH | -OCH2CH3 | -H | -H | -H |
| Compound 6 | -H | -OCH2CH3 | -OH | -H | -H |
| Compound 7 | -H | -H | -H | -NO2 | -H |
| Compound 8 | -H | -COH | -H | -H | -H |
| Compound 9 | -OCH3 | -H | -H | -OCH3 | -H |
| Compound 10 | -OCH3 | -H | -H | -H | -H |
| Compound 11 | -H | -OH | -H | -H | -H |
| Compound 12 | -H | -Br | -OH | -Br | -H |
2.3.2. Retrieval of Ligands and Screening
The structures of all curcumin analogs were obtained from the literature [17, 18] and used in molecular docking studies to investigate their interactions with the target protein. The reference and control compounds were retrieved from the PubChem compound database (https://pubchem.ncbi.nlm.nih.gov/) using PubChem CID (Curcumin: 969516, Tamoxifen: 2733526) [26]. Curcumin analogs were drawn and optimized by energy minimization using the Merck molecular force field (MMFF94) on the MarvinSketch application (https://www.chemaxon.com).
2.3.3. Molecular Docking Studies
AutoDock v4.2.6 software (AutoDock 4.2, Scripps Research, La Jolla, CA, USA, https: //autodock. scripps. edu/download -autodock4/, accessed on 20th December, 2023) was used in this study for molecular docking. Validation analysis of the native ligand was carried out to determine the Root Mean Standard Deviation (RMSD) value and grid box. RMSD showed 0.82 Å, and the grid box dimensions were adjusted on the X, Y, and Z coordinates to 50 × 44 × 46. The grid center was 31.767, -0.942, and 24.202, respectively, in the active site of Erα. AutoGrid was used to construct a 3D grid around the receptor's target binding location to make docking calculations easier [27]. The binding energy and inhibition constant of the interaction between the ligands and targets were estimated using the Lamarckian genetic algorithm (with a maximum of 100 conformers evaluated for each chemical to obtain optimal conformation). The remaining parameters were left as default for the molecular docking study.
AutoDock employs the Lamarckian Genetic Algorithm (LGA). This algorithm is used to determine the optimal docking position between the ligand and the protein, which then generates the function's empirical binding free energy value. This algorithm allows the ligand to move flexibly. Molecular mechanics components, including dispersion or repulsion, hydrogen bonding, electrostatic interactions, and covalent geometry optimization, constitute the first four terms in the algorithm's molecular free energy calculation. The final terms account for internal torsional strain, global rotation and translation, desolvation upon binding, and hydrophobic interactions. A good ligand conformation is typically associated with a lower binding free energy [28].
After the validation analysis and determining the algorithm, molecular docking was carried out between the curcumin analogs and the protein. All of the steps were similar to the validation analysis, including the dimensions of the grid box. The only difference was that we used our energy-minimized ligand for the analysis. The results showed the binding energy and interactions between the ligands and the protein, demonstrating their potential compatibility as anticancer agents.
3. RESULTS AND DISCUSSION
The curcumin analogs utilized in this study were dibenzylidene-cyclohexanone derivatives, which were successfully synthesized in a previous study and reported to have antioxidant activity better than tocopherol [17, 18]. Antioxidants help to protect cells from damage caused by free radicals or oxidative stress, which are one of the factors that can cause various diseases, including cancer [29]. It has been found that antioxidants like vitamin C (ascorbic acid), vitamin E (tocopherol), and beta-carotene have potential anti-cancer effects [30]. Therefore, our study investigated the potential of these curcumin analogs as anticancer agents, especially for the treatment of breast cancer.
The backbone structure (Fig. 1) with various substituents (Table 1) was designed using the Marvin Sketch application, generating 2D structures and canonical SMILES for each compound. These were used for virtual pharmacokinetic screening—including drug-likeness evaluation, pharmacophore modeling, and molecular docking. The primary target protein is human estrogen receptor alpha (ERα; PDB ID: 2IOG), which plays a key role in breast cancer.
3.1. Analysis of Drug-Likeness Properties
Drug-likeness properties and ADMET prediction were assessed using ADMETlab 3.0 [21]. Properties like molecular weight, hydrogen bond acceptor, hydrogen bond donor, and LogP were identified using Lipinski’s rule of five, as mentioned in Table 2. The molecule should have a molecular weight of ≤500, a partition coefficient (LogP) of ≤5, hydrogen bond donors of ≤5, and hydrogen bond acceptors of ≤10, respectively, to adsorb well following Lipinski's rule of five [20]. The LogP is a logarithm of the octanol-water partition coefficient, which measures a compound’s lipophilicity or its tendency to dissolve in lipids (like octanol) compared to water [31].
Oral bioavailability is believed to be significantly influenced by each of these characteristics, as well as molecular flexibility [32]. If the compound has two or more violations of Lipinski’s rule of five, its absorption in the intestine will be low. All curcumin analogs met Lipinski’s rule of five, except compound 12. Compound 12 did not meet the criteria in terms of molecular weight and LogP, indicating its poor oral bioavailability based on virtual screening.
3.2. Pharmacophore Modeling
Validation of the pharmacophore modeling method was carried out to confirm the ability of the pharmacophore model to distinguish the structures of active and inactive compounds [33]. Model nine showed the best performance with AUC values of 1.00, 1.00, and 0.85 at thresholds 1.5, 10, and 100%, respectively, along with 72 true positives (TP) and 32 false positives (FP). These values are very good as they are close to 1. The ROC plot is shown in Fig. (2A). The pharmacophore features are presented in Fig. (2B). Three features were created in the model: one hydrophobic feature (AR) depicted by a yellowish sphere, one hydrogen bond donor (HBD), and one hydrogen bond acceptors (HBA) seen with a red-green sphere shape.
| Compounds | Criteria | Drug-likeness | |||
|---|---|---|---|---|---|
| MW* (≤500) | HBA* (≤10) | HBD* (≤5) | LogP*(≤5) | ||
| Curcumin | Yes | 368.13 | 6 | 2 | 2.374 |
| Compound 1 | Yes | 489.98 | 3 | 0 | 5.01 |
| Compound 2 | Yes | 378.35 | 5 | 2 | 3.93 |
| Compound 3 | Yes | 356.43 | 1 | 0 | 5.03 |
| Compound 4 | Yes | 409.98 | 1 | 0 | 6.32 |
| Compound 5 | Yes | 394.18 | 5 | 2 | 4.07 |
| Compound 6 | Yes | 394.18 | 5 | 2 | 3.82 |
| Compound 7 | Yes | 364.11 | 7 | 0 | 4.40 |
| Compound 8 | Yes | 330.13 | 3 | 0 | 3.88 |
| Compound 9 | Yes | 394.18 | 5 | 0 | 4.65 |
| Compound 10 | Yes | 334.16 | 3 | 0 | 4.75 |
| Compound 11 | Yes | 306.13 | 3 | 2 | 3.63 |
| Compound 12 | No | 617.77 | 3 | 2 | 5.514 |
| Tamoxifen | Yes | 371.22 | 2 | 0 | 6.311 |
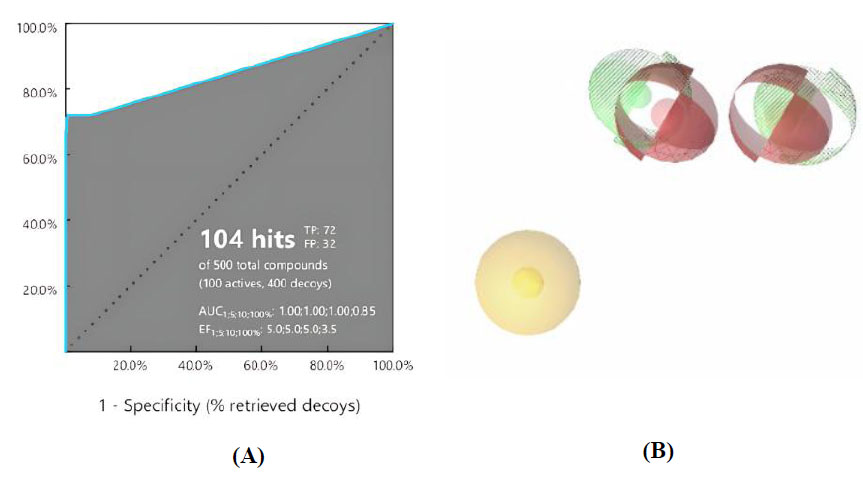
(A) ROC plot of the pharmacophore model and (B) Visualization of ligand-based pharmacophore features.
This pharmacophore model tested curcumin analogs mapped according to the same pharmacophore. From this study, it can be predicted that if the structures have the same pharmacophore, they will have the same biological properties [34]. The hit compounds are listed in Table 3, indicating that two out of twelve had the highest pharmacophore fit score. The pharmacophore fit-score values evaluated the degree of overlap between the pharmacophore features and the compound's chemical functions [23]. The pharmacophore study identified two curcumin analogs, compounds 4 and 11, that have pharmacophore fit-score values similar to the model and have the same pharmacophore as the active compound (training set).
| Compounds |
Pharmacophore Fit-Score |
Mapping in 2D and 3D |
|---|---|---|
| Model (active compound) | 46.87 | 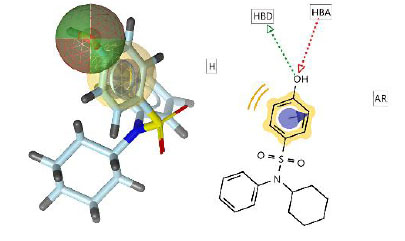 |
| Compound 4 | 36.87 | 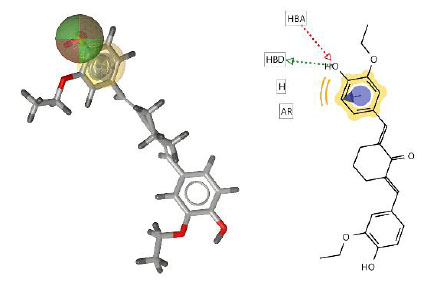 |
| Compound 11 | 36.65 | 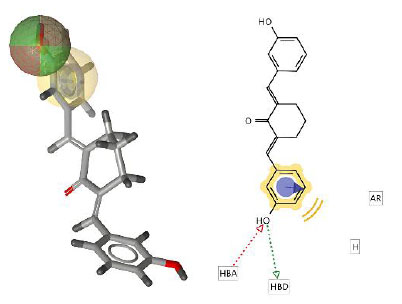 |
3.3. Molecular Docking
Molecular docking studies were conducted to predict the binding mode, affinity, binding free energy (ΔG), and interaction between a small molecule and protein target receptors [35]. They were carried out using AutoDock between ten curcumin analogs, curcumin, and tamoxifen against the ERα enzyme. Before molecular docking with the tested compounds, the validation method was performed first to determine the grid box and Root Mean Square Deviation (RMSD). RMSD was used to compare the docking orientation between the corresponding co-crystallized pose of the same ligand molecule. Three solutions were identified based on the RMSD value: (a) good solutions when the RMSD was less than 2.0, (b) acceptable solutions when the RMSD was between 2.0 and 3.0, and (c) bad solutions when the RMSD was greater than 3.0 [36].
The validation method result showed a good solution with an RMSD value of 0.82 Å. The interaction between the native ligand and the protein was based on literature data from the RCSB PDB, highlighting Asp351 as a key amino acid residue in the catalytic site. This residue was also identified from studies on the interactions of raloxifene and bazedoxifene with human ERα [25]. Subsequently, molecular docking of curcumin analogs was performed using the same validated method. In this study, curcumin was used as a lead compound, and tamoxifen was used for comparison. The binding energy and inhibition constant (Ki) of curcumin analogs are presented in Table 4.
| Compounds | Binding Energy (kcal/mol) | Inhibition Constant (Ki) (nM) | 2D Interaction (Protein-ligand) |
|---|---|---|---|
| Native ligand (Compound 11F) | -14.51 | 0.02 | 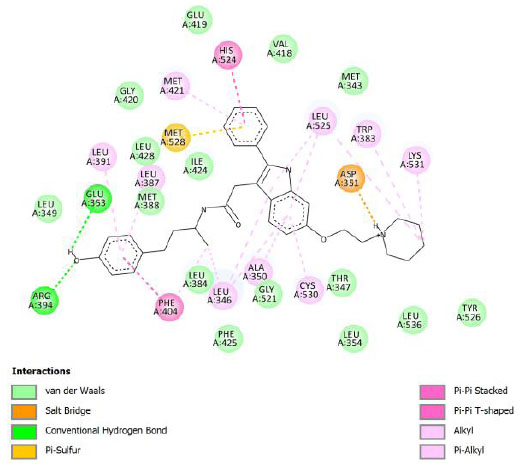 |
| Curcumin | -9.18 | 186.65 | 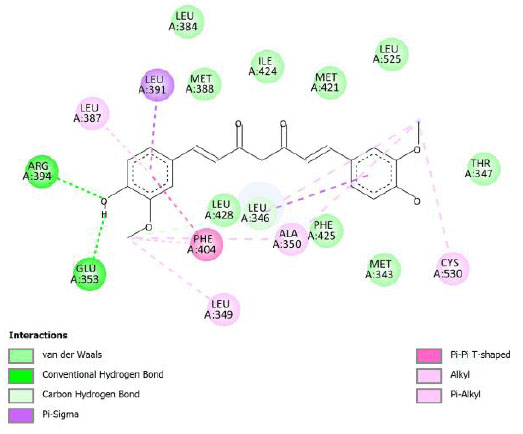 |
| Compound 1 | -10.76 | 12.87 | 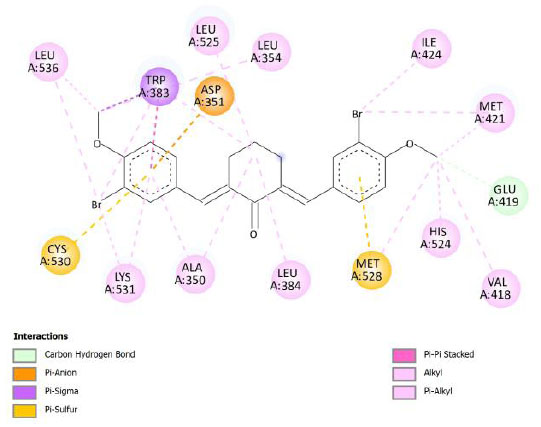 |
| Compound 2 | -9.22 | 174.44 | 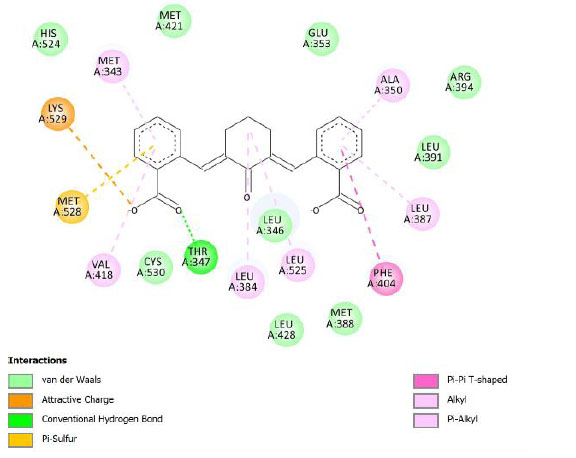 |
| Compound 3 | -10.09 | 40.08 | 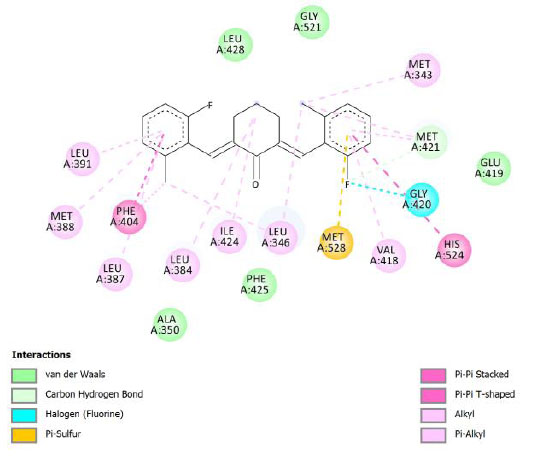 |
| Compound 4 | -11.10 | 7.36 | 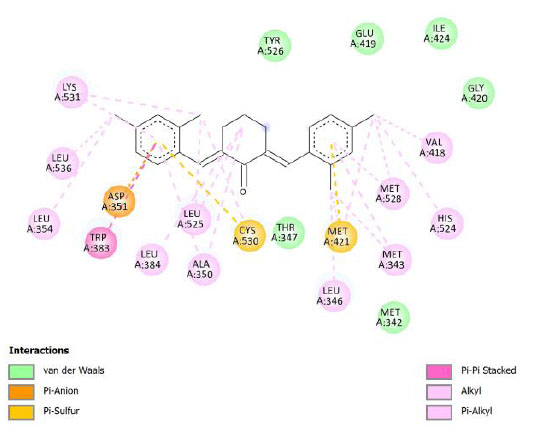 |
| Compound 5 | -9.29 | 155.92 | 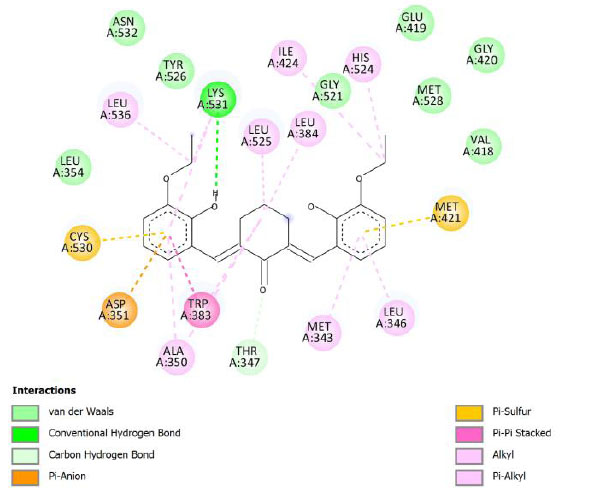 |
| Compound 6 | -10.24 | 31.38 | 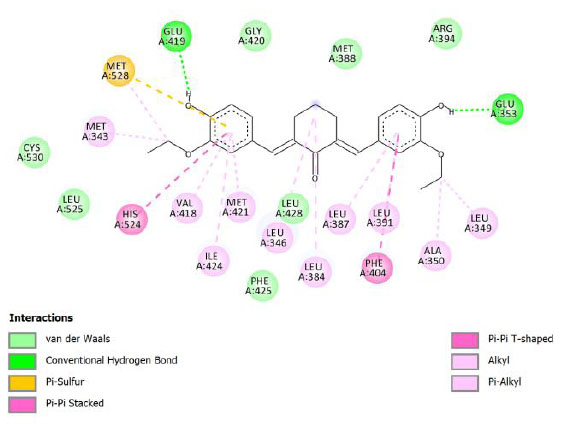 |
| Compound 7 | -10.24 | 31.12 | 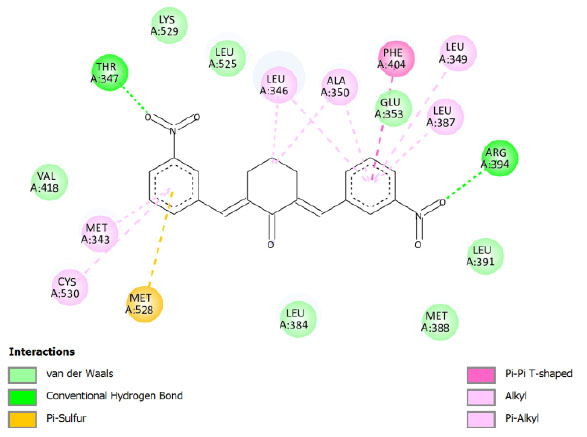 |
| Compound 8 | -10.17 | 34.88 | 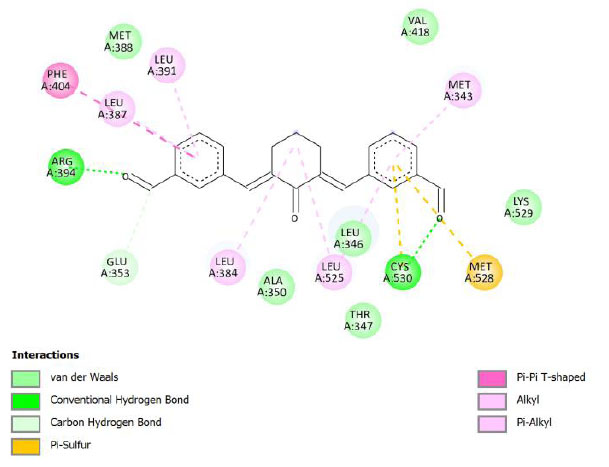 |
| Compound 9 | -9.80 | 65.82 | 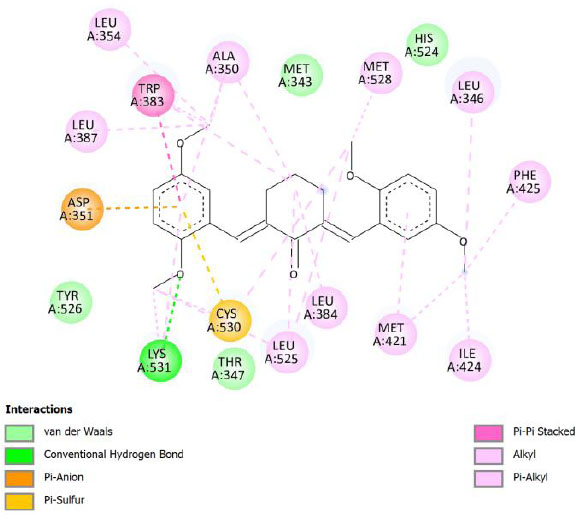 |
| Compound 10 | -9.77 | 68.67 | 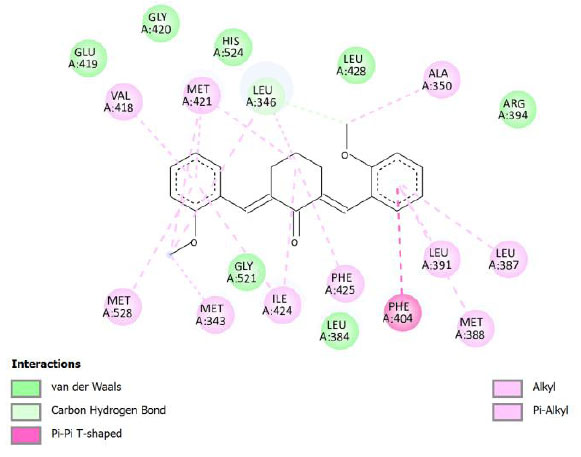 |
| Compound 11 | -9.97 | 49.07 | 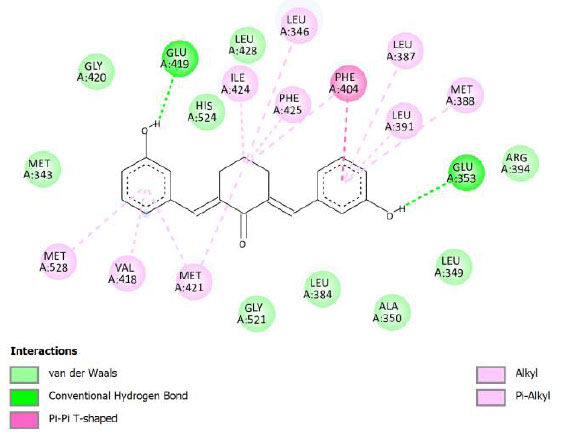 |
| Compound 12 | -12.06 | 1.45 | 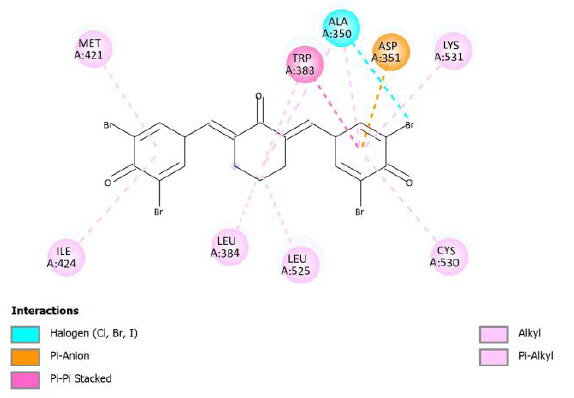 |
| Tamoxifen | -10.45 | 21.87 | 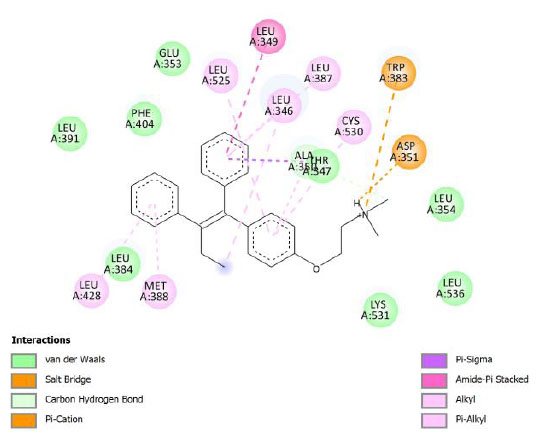 |
Tamoxifen is a common treatment for breast cancer; it works by reducing estrogen activity in certain tissues. Similar to many cancer drugs, it has various adverse effects. Hot flashes, irregular periods, vaginal discharge, peripheral edema, hypertension, mood swings, discomfort, depression, skin rashes and changes, weakness, nausea and vomiting, arthritis, arthralgia, lymphedema, and pharyngitis are some of the other frequent side effects caused by tamoxifen [37]. There is a need to search for drugs that have lower side effects to overcome the increasing drug resistance.
Binding energy refers to the strength of the interaction between a biomolecule and its ligand. The dissociation constant of the enzyme-inhibitor combination is represented by the Ki value, also known as the inhibition constant. A decreased chance of dissociation is indicated by a lesser Ki value, which, in turn, leads to stronger inhibition [38]. As mentioned in Table 4, the best binding energy was demonstrated by compound 12, followed by compound 4 and compound 1, with values of -12.06, -11.10, and -10.76 kcal/mol, respectively, better than tamoxifen and curcumin as existing drug and lead compound. They showed interactions with important residues with the Asp351. The interactions and the conformation are presented in Table 4.
Compound 12 demonstrated the highest binding energy than other curcumin analogs. It has substituent -Br in R2 and R4 and -OH in R3. The key residue, Asp351, interacted with the side aromatic structure in compound 12 with the π-anion type of interaction. π-Anion interaction is one non-covalent interaction that can build between a negatively charged species (anion) and the electron-deficient π system of an aromatic ring [39]. In addition, its bonding energy is supported by several other interactions, such as halogen interaction and π-alkyl interactions. The π-alkyl interaction is an interaction that occurs in a hydrophobic environment where aromatics and alkyl sides can interact (part of the van der Waals interaction) [40]. Compound 12 was found to be a complex compound as it demonstrated poor pharmacokinetics profiles, based on Lipinski’s rule of five. However, some research works have indicated that formulations can improve the bioavailability or pharmacokinetics profile of drugs, like nanosuspensions, solid lipid nanoparticles, liposomes, or microemulsions [41].
In compound 4 and compound 1, with -Cl substituents in R1 and R3 (compound 4) and -Br in R2 and -OCH3 in R3 (compound 1), Asp351 residues interacted with aromatic structures forming π-anion interaction, similar to compound 12. While compound 11F acted as a native ligand, Asp321 interacted with the H group in the cyclohexanone to form a salt bridge interaction. Salt bridges are ionic interactions between oppositely charged groups. Salt bridges tend to be stronger than π-alkyl interactions [40]. Other interactions that occurred between compound 4 and protein were van der Waals, π-anion, π-sulfur, π-π, and alkyl interactions. In compound 1, there were carbon-hydrogen bonds, π-anion, π-sulfur, π-π, and alkyl interactions that contributed to binding energy.
The curcumin analogs have higher binding energy than curcumin and tamoxifen but less than the native ligand. There is a conventional hydrogen bond in the interaction between native ligand and protein. This interaction plays a crucial role in drug binding [42], which likely explains why the native ligand exhibited a higher binding energy.
The development of artificial intelligence (AI) tools and technology has made AI-driven drug discovery more prominent. It offers speed, cost-effectiveness, and the ability to identify novel drug candidates and repurpose existing ones, whereas traditional methods like molecular docking can be computationally intensive and may struggle with large-scale screening [43]. However, we found that traditional molecular docking is better than AI-driven drug discovery, such as deep learning. This is because some of the deep learning approaches employ an entire protein (blind docking) for docking, which does not satisfy the common requirements, whereas the old method uses the catalytic site as the binding site [44].
Many advanced deep learning techniques, such as those used for low-dose CT image denoising, could enhance the quality of this study. Improved imaging quality would enable more accurate visualization of tumor morphology and the response to curcumin-based drug candidates. Additionally, high-resolution imaging could support the identification of subtle biomarkers that are essential for understanding the mechanisms of action of medications based on curcumin and their influence on breast cancer progression. Furthermore, the integration of advanced techniques, such as attention-guided enhanced U-Net architectures, offers the opportunity to develop robust predictive models that combine imaging data with molecular and pharmacological information, facilitating a more comprehensive and data-driven approach to drug development [45, 46].
CONCLUSION
This study is a computational study that aims to explore dibenzylidene-cyclohexanone curcumin analogs as anticancer agents for the treatment of breast cancer. It analyzed drug similarity based on Lipinski's rule of five using ADMETlab 3.0 (virtual screening), then explored structural similarity based on pharmacophore modeling using LigandScout, and finally determined how the curcumin analogs interact with hERα receptor based on molecular docking. The virtual screening revealed that all of the curcumin analog compounds met the criteria of Lipinski’s rule of five, except 12 because it has a LogP of more than 5 and a molecular weight of more than 500 Da. Pharmacophore modeling results showed that compounds 4 and 11 have similar structures to compounds that have activity as hERα inhibitors (training compounds) with high pharmacophore fit-score, while molecular docking results revealed that curcumin analog compounds 12, 4, and 1 have better binding energy values (-12.06, -11.10, and -10.76 kcal/mol, respectively) than curcumin (-9.18 kcal/mol) and tamoxifen (-10.45 kcal/mol), one of the anticancer drugs. Research on curcumin analogs as potential inhibitors of breast cancer proteins warrants further investigation. Future studies are expected to include molecular dynamics simulations, as well as in vitro and in vivo experiments, to thoroughly evaluate the potential and mechanisms of these curcumin analog compounds.
LIST OF ABBREVIATIONS
| ADMET | = Absorption, Distribution, Metabolism, Excretion, and Toxicology |
| AUC | = Areas Under the Curves |
| BE | = Binding Energy |
| EF | = Enrichment Factors |
| HBA | = Hydrogen Bond Acceptors |
| HBD | = Hydrogen Bond Donors |
| HBD | = Hydrogen Bond Donors |
AVAILABILITY OF DATA AND MATERIALS
All the data and supporting information are provided within the article.
FUNDING
This work was supported by the Directorate General of Higher Education, Research, and Technology, Ministry of Education, Culture, Research, and Technology, Indonesia, through the Pendidikan Magister menuju Doktor untuk Sarjana Unggul (PMDSU) (https://www.pmdsu.id/) research scheme with grant no. 048/E5/PG.02.00.PL/2024 and contract no. 2790/UN1/DITLIT/PT.01.03/2024. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
ACKNOWLEDGEMENTS
The authors would like to express their gratitude to the Department of Medicinal Chemistry and Analysis, Faculty of Pharmacy, Padjadjaran University, especially Prof. Apt. Muchtaridi, Ph.D., for generously lending the computer and license for pharmacophore modeling (LigandScout). The contributions have enhanced the quality of this research. The authors would also like to acknowledge the bioinformatics tools, software, and databases (ADMETlab 3.0, MarvinSketch, ChemDraw, Autodock v4.2.6) utilized in this work.

