
FastImpute: Development and Validation of a Workflow for Open-source, Reference-Free Genotype Imputation Methods - An Example in Breast Cancer (PRS313_BC)
Authors Info & Affiliations
Abstract
Background
Genotype imputation is crucial for enhancing genetic data from genotyping arrays by predicting missing single nucleotide polymorphisms (SNPs). Traditional imputation methods often compromise data privacy or are computationally demanding, limiting their accessibility. While newer deep learning methods offer a privacy-preserving alternative, their large model sizes make them difficult to deploy on client-side devices like personal computers or smartphones.
Methods
We developed FastImpute, a workflow for creating lightweight, reference-free imputation models designed for client-side deployment. As a case study, we trained linear and logistic regression models to impute SNPs for the breast cancer polygenic risk score, PRS313_BC. We used whole-genome sequencing data from 2,504 individuals in the 1000 Genomes Project as a training and testing set. The models were trained to predict target PRS SNPs using input from SNPs on commercial genotyping arrays. Performance was evaluated against true sequencing data and benchmarked against Beagle.
Results
The correlation (R2) between a PRS calculated using our simple linear regression model and a PRS calculated using true sequencing data was 0.86. This significantly outperformed both no imputation and simple minor allele frequency imputation (R2 = 0.38). Our lightweight models performed comparably to Beagle in identifying high-risk individuals, correctly classifying 3 (linear) and 4 (logistic) out of 6 individuals in the top 1% of risk, similar to Beagle (4 out of 6).
Conclusion
The FastImpute pipeline demonstrates that simple, lightweight models can provide effective and privacy-preserving, and accessible genotype imputation, enabling real-time genetic risk assessment on edge devices.
Availability
Web application: https://aaronge-2020.github.io/FastImpute/
Code: https://github.com/aaronge-2020/FastImpute
1. INTRODUCTION
Genotype imputation enhances genetic data by predicting missing single nucleotide polymorphisms (SNPs) using reference haplotype information [1, 2]. Traditional methods leverage linkage disequilibrium (LD), inferring untyped single nucleotide polymorphism (SNP) genotypes by assuming similar LD structures between genotyped target sets and fully sequenced reference panels [2]. However, these methods often rely on external services like the Michigan Imputation Server [3], which can compromise data privacy or require downloading entire reference genomes, which is computationally inefficient.
Recently, deep learning-based methods [4, 5, 6, 7], utilizing advanced architectures such as Transformers and Recurrent Neural Networks, have emerged as a promising alternative, representing the state-of-the-art for raw imputation accuracy. These methods predict missing genotypes using pre-trained models, enhancing privacy and accessibility. This approach aligns with the increasing preference for FAIR computational solutions (findable, accessible, interoperable and reusable) in epidemiology [8, 9]. (We refer to these pre-trained models as “reference-free methods,” since they do not require end users to download reference genomes locally or upload data to an external server housing reference genomes. We stress, however, that these models are trained on a set of reference genomes, even if the reference genomes are not required for model deployment.)
Despite their promise, previous reference-free methods face a critical limitation; however, the high accuracy of these state-of-the-art models comes with a significant trade-off in computational cost. They often target specific genomic regions, such as the major histocompatibility complex (MHC) region, due to its high degree of polymorphism and structural variation [10]. Their large model sizes and computational intensity make them inefficient or unsuitable for client-side deployment. Furthermore, retraining these models for different regions requires substantial computational resources.
The accompanying open-source in-browser application, FastImpute, addresses these limitations by providing a baseline for zero-footprint client-side imputation methods. Our pipeline produces models that can be implemented using web technologies, primarily coded in JavaScript, leveraging advanced computational resources available in modern web browsers. This approach, including access to libraries like TensorFlow via Web Assembly, has been demonstrated to be feasible for estimating cancer risk in user-facing applications [11, 12].
2. METHODS
2.1. Study Design
This study employed a quantitative, observational study design to develop and validate a computational pipeline, FastImpute. The research involved gathering publicly available genomic data, selecting relevant SNP subsets, training predictive models, and evaluating their performance against a gold standard and an established imputation tool. The primary research goal was to create a lightweight, client-side imputation method and assess its accuracy for calculating polygenic risk scores.
2.2. Data Source and Sample Size
The primary dataset was the Whole Genome Sequencing (WGS) data from the 1000 Genomes Project (Phase 3, GRCh37), which includes 2,504 individuals from 26 diverse populations. The samples for the 1000 Genomes Project are anonymous and do not have associated phenotypic or medical data. More details can be found in the original paper [13]. To define the SNP panel for a common direct-to-consumer (DTC) platform, we analyzed 119 23andMe V5 chip data files from users who made their data public on OpenSNP.org [14]. It is important to note the distinct roles of these datasets: the large and diverse 1000 Genomes Project data was used for all model training and validation, while the 119 23andMe files were used solely for the technical purpose of defining a representative list of input SNPs found on the consumer chip. For model development, the 1000 Genomes Project data was randomly split into a training set of 2,003 individuals (80%) and a testing set of 501 individuals (20%).
2.3. The FastImpute Pipeline
The FastImpute pipeline provides a complete workflow for creating the lightweight, reference-free, and privacy-preserving imputation models that are central to this work. This versatile pipeline is designed to predict various genomic regions across different genotyping chips. To showcase its application in detail, we used the calculation of a polygenic risk score for breast cancer, PRS313_BC (PGS Catalog entry PGS000004) [15], using genotyping data available on a commercial genotyping chip, the 23andMe V5 Gene Panel [16], as a running case study throughout the paper. PRS313_BC comprises 313 SNPs used for breast cancer risk prediction, most of which are not present on the V5 gene chip and must be imputed from nearby observed genotypes.
As shown in Fig. (1), the FastImpute pipeline comprises four key steps:
- Gather Whole Genome Sequencing (WGS) data (e.g., from the 1000 Genomes Project [13]).
- Subset the panel SNPs from WGS data to include only those present on the input genotyping platform (in our example, the 23andMe V5 Gene Panel).
- Use LDlink [17] to identify a subset of SNPs in the raw input that are in LD with the target SNPs (PRS313).
- Train the model to predict the target SNPs using the input SNPs.
To establish a baseline, we trained two models: a logistic regression model on phased data and a linear regression model on unphased data. We deployed the linear regression model, which processes unphased data, using web technologies [18, 19] (Web-stack). This approach offers superior reusability and privacy compared to native applications, enhancing user data protection by processing all information locally within the browser.
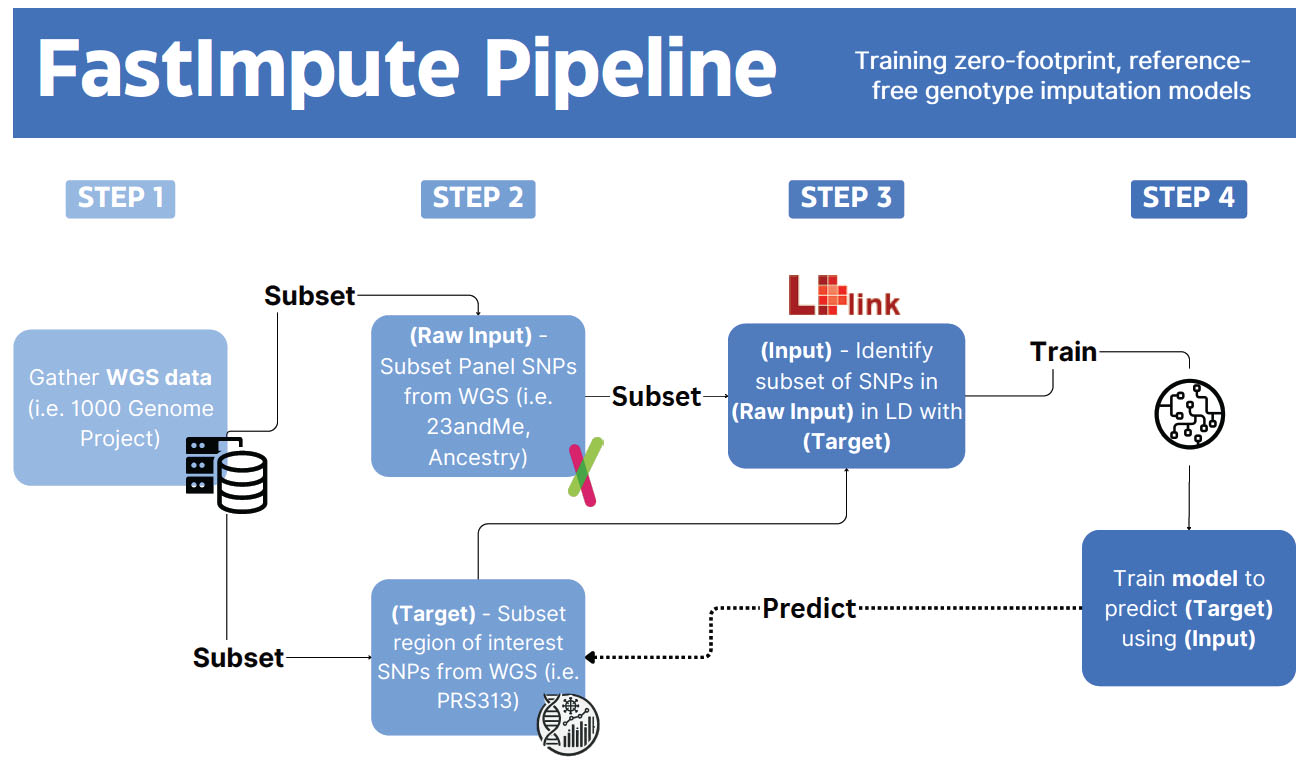
The FastImpute pipeline illustrated with PRS313_BC as an example. The pipeline comprises four key steps. Step 1: Gather Whole Genome Sequencing (WGS) data (e.g., from the 1000 Genomes Project). Step 2: Subset the panel SNPs from WGS data to include only those present in platforms like 23andMe and AncestryDNA, and a region of interest like PRS313. This subset serves as the raw input and target (output) for the model. Step 3: Using LDlink, identify a subset of SNPs in the raw input that are in linkage disequilibrium (LD) with the target SNPs (PRS313). These SNPs in LD with the target SNPs serve as the input for model training. Step 4: Train the model to predict the target SNPs using the input SNPs.
3. RESULTS
The implementation of FastImpute will be described here in detail for imputing the SNPs of PRS313_BC [15] from SNPs available on the 23andMe V5 chip.
3.1. Preparing the Data: Determining the 23andMe SNPs on the V5 SNP Chip
We filtered 23andMe files generated from 2022 onwards on OpenSNP [14], ensuring they contained between 600,000 and 700,000 positions and shared at least 60% of SNPs with a reference V5 chip file. This process yielded 119 23andMe files. Due to quality control measures, SNPs sampled from the same V5 chip can vary slightly, necessitating a method to ensure consistency across different datasets. Consequently, as shown in Fig. (2), while 70 out of the total 77 PRS313_BC SNPs present in the 23andMe chip are found in over 75% of the user data, there are 7 PRS313_BC SNPs that appear only sporadically.
3.2. Steps 1, 2, and 3: Preparing the Training Dataset
We downloaded the 1000 Genomes Project Data GrCh37 (Step 1, Fig. 1) and subsetted the PRS313_BC SNPs and the 23andMe panel SNPs (Step 2, Fig. 1). Using LDProxy, with all populations of the 1000 Genomes Project as the reference panel, we obtained LD data for each PRS313_BC SNP (Step 3, Fig. 1), focusing on 23andMe SNPs with an R2 value greater than 0.01. For multiallelic SNPs not found in NCI's LD Proxy service, we included all SNPs within a 500K base pair window, resulting in 17,551 positions used for training and evaluation.
We processed these positions to retrieve allele dosages, converting multiallelic variants to binary format. We created two versions of this data: one summing allele dosages to simulate unphased data, and another maintaining the phased data format.
3.3. Step 4: Model Training
Since the models are designed to capture LD patterns, inter-chromosomal information is unnecessary for predicting SNP dosages. Therefore, we split the 23andMe panel data by chromosome, allowing us to construct separate models for each chromosome (excluding X and Y, since they are not a part of PRS313). Hence, we developed 44 models: 22 logistic regression models for phased data and 22 linear regression models for unphased data.
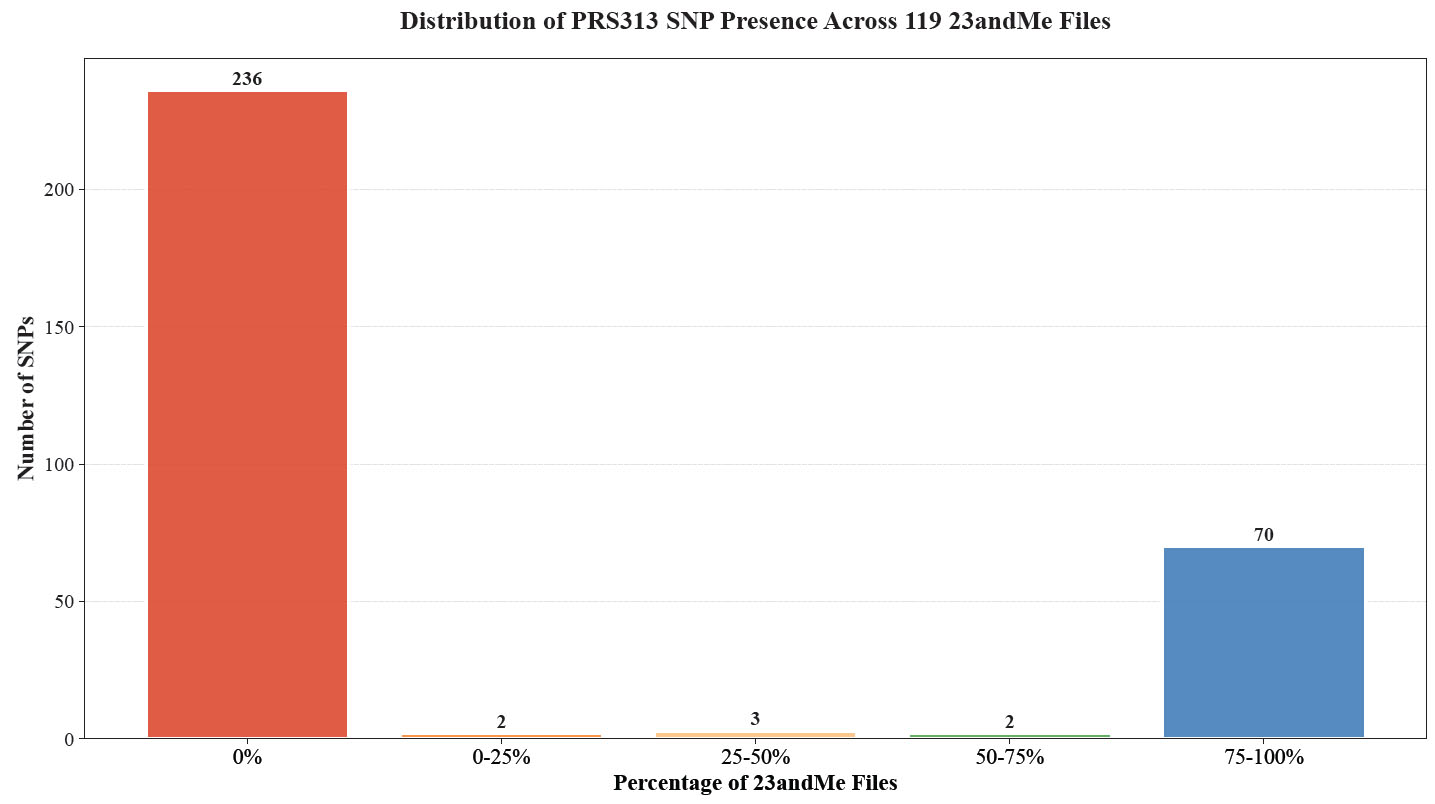
Distribution of PRS313_BC SNP Presence Across 119 23andMe Files from OpenSNP [14]. The bar chart shows the distribution of the presence of PRS313_BC SNPs across 119 23andMe V5 chip files. In total, 77 SNPs were found to be present within the 23andMe V5 chip. The x-axis represents the percentage of 23andMe files containing each SNP, divided into five ranges: 0%, 0-25%, 25-50%, 50-75%, and 75-100%. The y-axis indicates the number of SNPs within each range. A significant number of SNPs (236) are not present in any of the files (0%), while 70 SNPs are present in 75-100% of the files. The other ranges (0-25%, 25-50%, and 50-75%) contain very few SNPs, with counts of 2, 3, and 2, respectively. This distribution highlights the variability and inconsistency in SNP presence across different 23andMe V5 chip datasets.
Each of these models was trained to impute all PRS313_BC SNPs on their respective chromosome, regardless of whether the SNP was present on the V5 chip or not. This was done to ensure that any 23andMe user could get a PRS score regardless of which SNPs on the chip were missing. For each target PRS313_BC SNP on a given chromosome, we used all the V5 SNPs on that same chromosome that were in LD (R2 > 0.01 via LDProxy) with the target SNP as input features to train the prediction model. Since most of the SNPs in the V5 chip were not in LD with the PRS313_BC SNPs, they were not used as input to our model. These models were implemented using PyTorch [20] and included L1 regularization [21] to prevent overfitting.
The logistic regression models output probabilities for each allele, which were thresholded at 0.5 to assign binary predictions (0 or 1) for each chromosome. These binary predictions were then summed to derive discrete genotypes (0, 1, or 2). In contrast, the linear regression models directly predicted continuous dosage values (ranging from 0 to 2), which were rounded to the nearest integer to assign discrete genotypes. For polygenic risk score (PRS) calculation, we used the “best-guess” genotypes (rounded dosages) rather than allele dosage (expected allele count based on posterior probabilities). Both models ultimately converted probabilities or continuous dosages into discrete genotypes for PRS calculation.
We used an 80/20 simple random data split for training (n=2003) / testing (n=501) and employed Optuna [22] for hyperparameter tuning with 10-fold cross-validation across 50 trials for each chromosome.
3.4. Benchmarking Beagle
To benchmark the performance of Beagle 5.4 [23], we left out the same 501 samples that we used to evaluate our previously trained models from the 1000 Genome Project to serve as the test set. We then ran Beagle 5.4 on the full 23andMe panel data, excluding the overlapping PRS313_BC SNPs that are already present in the panel, using the remaining 2003 samples from the 1000 Genomes Project as the reference genome.
3.5. Deployment
We deployed our linear regression (unphased) model on GitHub at https://aaronge-2020.github.io/FastImpute/, enabling users to conveniently and privately calculate their PRS313_BC scores on any device, including smartphones (Fig. 3).
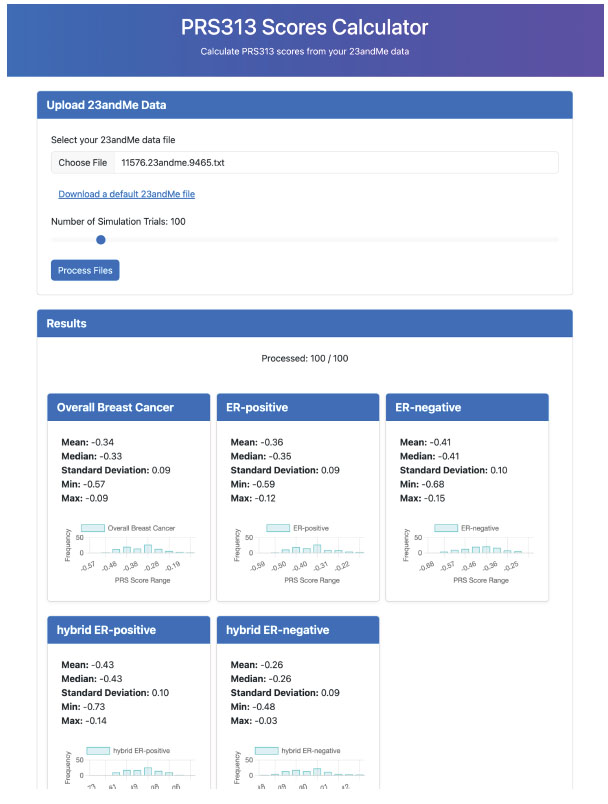
The PRS313_BC Scores Calculator interface. This website displays the results of the PRS calculations for various breast cancer phenotypes based on user's 23andMe genotype data. Users can upload their 23andMe data file and specify the number of simulation trials to process their PRS313_BC scores. The results section displays the processing status and the calculated PRS scores for five breast cancer phenotypes: Overall Breast Cancer, ER-positive, ER-negative, hybrid ER-positive, and hybrid ER-negative. Each phenotype panel includes statistical summaries of the PRS scores, such as mean, median, standard deviation, minimum, and maximum values, along with a histogram showing the distribution of PRS scores across the simulation trials. For more information, please see section 2.4.
To calculate the PRS from a user's 23andMe genotype data, we first converted the data from genotypes to allele dosages using the 1000 Genomes Project [13] as a reference for alleles at each position. Due to varying missing data across users, we imputed missing input array SNP values using simulations, taking independent draws based on minor allele frequency (MAF) data from the 1000 Genomes project. This imputation of missing V5 chip SNPs was performed before applying the imputation models to ensure complete input data. We then calculated PRS313_BC risk scores across multiple simulations, generating a distribution of potential risk scores. Users can specify the number of simulations, balancing accuracy and computational time.
For each simulation, two random draws were conducted for each allele, with the probability based on the MAF, to determine the dosage. The simulated data was then passed into the imputation model to impute the PRS313_BC SNPs. We used the imputed dosages (or actual dosages for already genotyped locations) to calculate PRS scores. Beta values for each SNP, corresponding to different breast cancer phenotypes, were retrieved from an external dataset. PRS scores were computed by multiplying the imputed dosages by their respective beta values and summing these products for each phenotype.
Results from these simulations were aggregated, and statistical summaries (mean, median, standard deviation, minimum, and maximum values) were computed for each phenotype. Finally, results were visualized using binned histograms to display the distribution of PRS scores, providing a variance estimate for the PRS score. This process typically takes less than 10 seconds for 1,000 simulations, though time may vary based on the user's machine.
3.6. Evaluation
The performance of different genotype imputation methods was evaluated at the SNP level using R2 (coefficient of determination) between the imputed and actual allele count in the testing data, imputation quality score (IQS) [15], area under the receiver operating characteristic curve (AUC), and accuracy to determine their effectiveness in genotype imputation and predicting PRS. R2 was calculated using the Scikit-learn [24] library to indicate the proportion of variance in the true genotypes explained by the imputed dosages. AUC was computed using PyTorch [20] to assess the discriminative ability of the imputed dosages. Our analysis, presented in Figs. (4 through 7), has shown that although Beagle [23] consistently performed the strongest across various metrics and PRS phenotypes, the baseline linear models do not fall significantly behind.
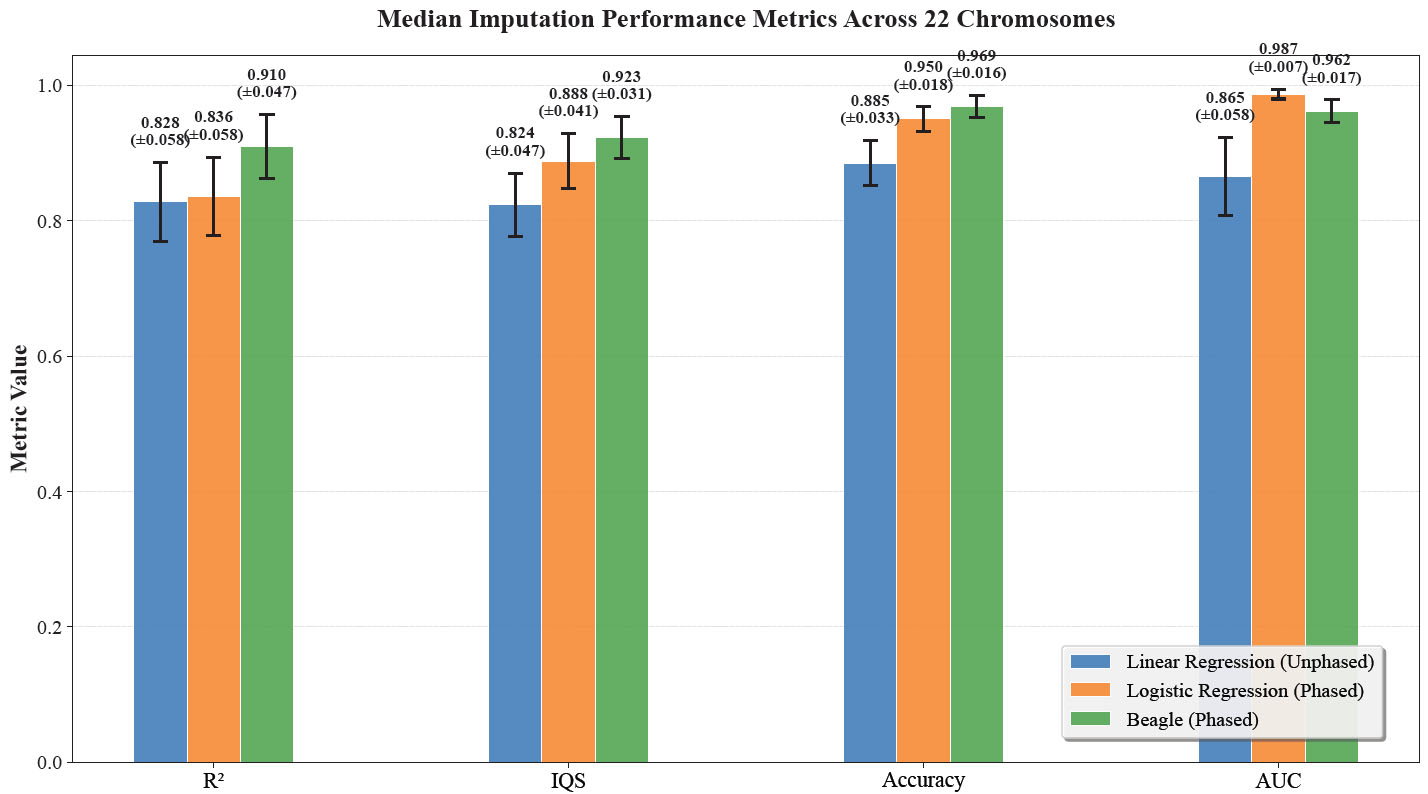
Median PRS313_BC Imputation Metrics Across 22 Chromosomes for Different Methods. These bar plots display the median evaluation metrics (R2, IQS, Accuracy, and AUC) across 22 chromosomal models for three genotype imputation methods: Linear Regression trained on unphased data, Logistic Regression trained on phased data, and Beagle using phased data. The metrics for each chromosome model were first calculated, and then the median values across all 22 chromosomes were determined. Error bars represent the standard deviation of the metrics across the different chromosomes.
3.6.1. Evaluation of Genotype Imputation Models
Since imputing allele dosages is a multi-class classification problem (with allele dosages of 0, 1, or 2), we calculated the one-vs-all AUC for each class for the linear regression model trained with unphased data. We then computed the mean AUC of the three classes. In cases where there were no positive classes for a genotype, resulting in an undefined AUC value, these undefined values were excluded when calculating the mean AUC.
For each chromosome, the model was evaluated on its corresponding test set, and performance metrics were calculated by aggregating predictions across all SNPs (micro-averaged for classification metrics like AUC-ROC). The median values of these chromosome-level metrics across all 22 chromosomes are reported in Fig. (4). As shown in Fig. (4), Beagle consistently achieves the highest median values. When assessed using IQS, accuracy, and AUC, logistic Regression (phased) and linear Regression (unphased) perform comparably to Beagle. While the Logistic Regression (phased) achieved a median IQS of 0.888 +/- 0.041 and the Linear Regression (unphased) had an IQS of 0.824 +/- 0.047, Beagle has a median IQS of 0.923 +/- 0.031. However, when assessed using R2, Beagle performs much stronger than the linear methods, with an R2 of 0.910 +/- 0.047, compared to 0.828 +/- 0.058 of logistic regression and 0.836 +/- 0.058 of linear regression.
The R2 of each individual SNP within PRS313_BC was computed for both the linear regression and logistic regression models. The distributions of these R2 values are plotted in Fig. (5) and compared with the R2 values of Beagle. This distribution further illustrates the performance differences between imputation methods. Beagle exhibits a high proportion of SNPs with R2 > 0.9, significantly outpacing Linear Regression (unphased), which only achieves this threshold for 18.53% of SNPs. Similarly, logistic Regression (phased) reaches this level for 31.57% of SNPs, highlighting its stronger performance over the unphased model.
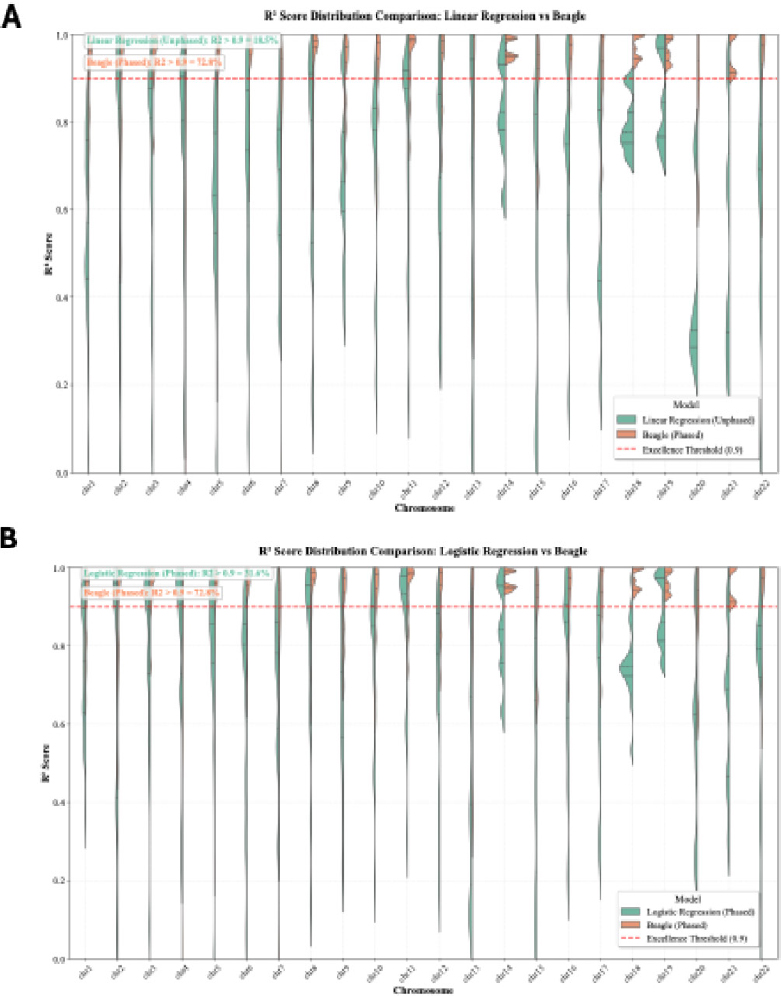
R2 Score Distributions for SNP Genotype Imputation Models across Chromosomes. 5A shows the R2 scores of SNPs imputed using the Linear Regression model trained on unphased data vs. Beagle results imputed using phased data. The violin plots illustrate the distribution across 22 chromosomes. The Beagle model shows a higher proportion of SNPs with R2 > 0.9 (72.84%) compared to the Linear Regression model (18.53%). 5B shows the R2 scores of SNPs imputed using the Logistic Regression trained on phased data vs. Beagle results imputed using phased data. Similar to (A), the Beagle model outperforms the linear method, with 72.84% of SNPs exceeding R2 > 0.9, compared to 31.57% for Logistic Regression.
3.6.2. Evaluation of PRS Scores Accuracy with Imputed Genotypes
The R2 values, depicted in Fig. (6), compare PRS scores calculated using imputed input genotypes to those calculated with real genotypes. Beagle achieves the highest R2 values across all phenotypes, with an R2 of 0.95 for overall breast cancer, indicating a strong correlation between imputed and actual genotypes. However, it is noteworthy that the baseline models—Logistic Regression (phased) and Linear Regression (unphased)—also perform surprisingly well. For instance, Logistic Regression achieves an R2 of 0.86 for overall breast cancer, only slightly lower than Beagle. These models significantly outperform the PRS score calculated without imputation and the Imputation with MAF method, both with an R2 of 0.38.
The confusion matrices in Fig. (7) illustrate the agreement between true and predicted quantiles for overall breast cancer risk using PRS313_BC scores. As shown in Fig. (7), out of the six test-set subjects in the top 1% of PRS scores, both the logistic regression model and Beagle correctly classified 4 out of 6. The linear regression model correctly classified 3 out of 6, while imputing with MAF only led to 1 out of 6 being correctly classified. For the misclassified patients in the top 1% of PRS scores, Beagle, linear regression, and logistic regression placed all of them in the next highest score quantile (1-5%). In contrast, imputing with MAF placed 2 out of 6 patients in the 5-10% quantile and 1 out of 6 in the 10-20% quantile.
The difference in performance becomes even more apparent in the 1-5% quantile, where imputing with MAF correctly classified only 4 out of 20 patients, with almost half (8 out of 20) falling outside the top 20% quantile. Beagle correctly classified 15 out of 20, and both linear and logistic regression models correctly classified 12 out of 20, with misclassified samples mostly found within adjacent quantiles.
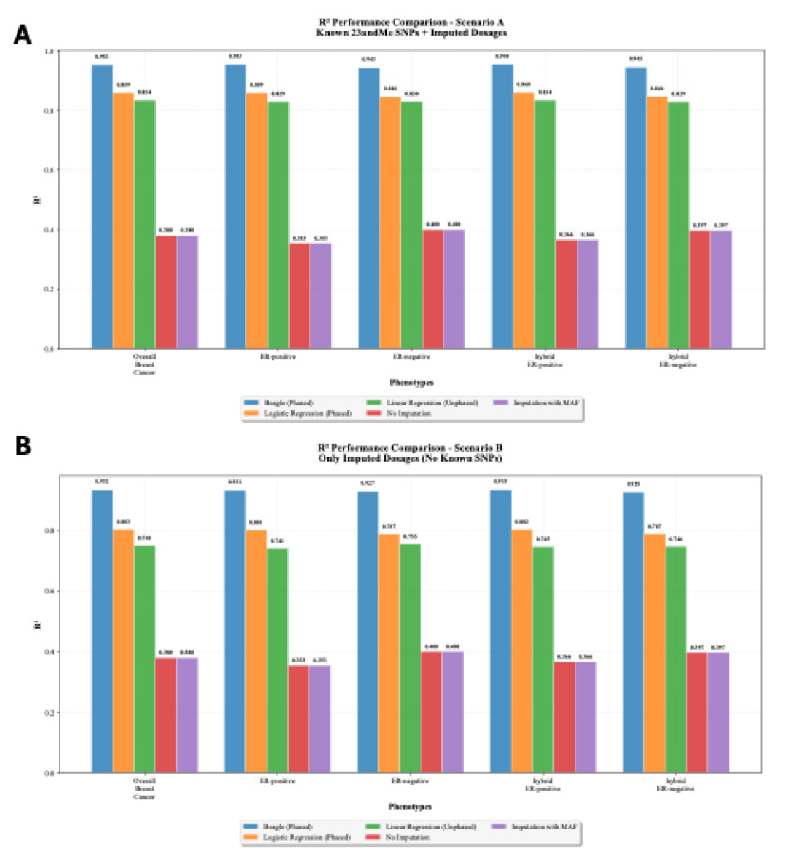
Comparison of the R2 of the PRS score Across Different Models. The R2 values were calculated by comparing the PRS scores calculated using imputed genotypes versus the PRS scores calculated using the real genotypes obtained from the WGS data. Due to quality control measures, although there are 77 SNPs present within the 23andMe genotyping panel, user data may have varying numbers of PRS313_BC. (A) R2 values when the PRS scores are calculated using all 77 known SNP positions and imputed dosages for the remaining SNPs. (B) R2 values when the PRS scores are calculated using only imputed dosages (assuming the user data has none of the PRS313_BC SNPs). The difference between (A) and (B) highlights the impact of the imputation process on the PRS calculation accuracy.
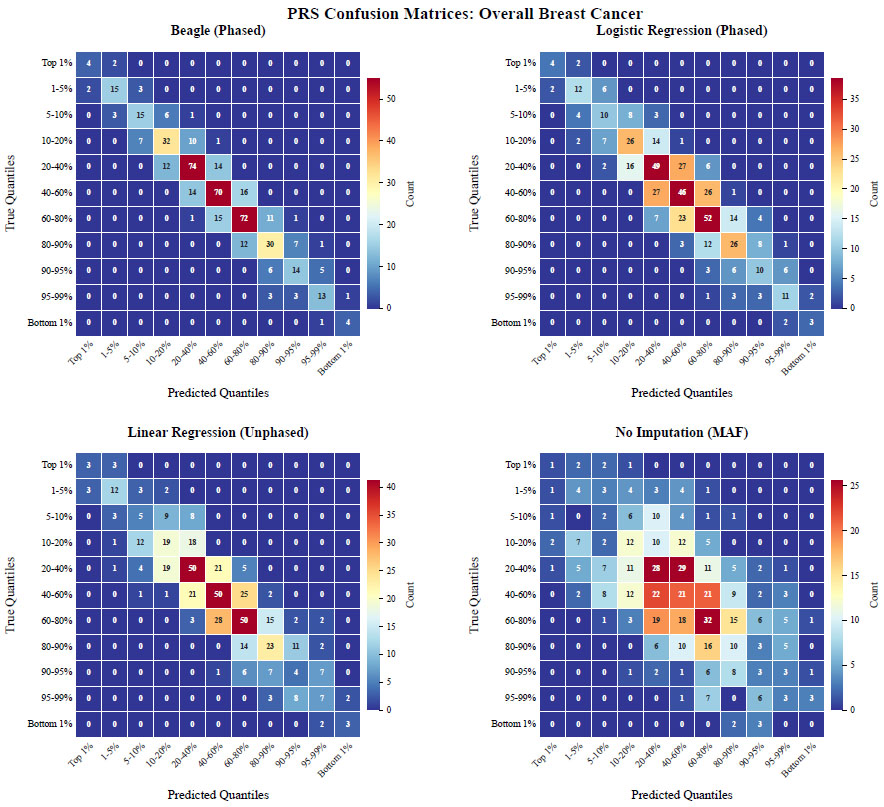
Confusion Matrices for Overall Breast Cancer Risk Prediction. The figure shows confusion matrices comparing the performance of different imputation methods for predicting overall breast cancer risk using PRS313_BC scores. The matrices display the agreement between true quantiles and predicted quantiles across various imputation techniques. Each confusion matrix's x-axis represents the predicted quantiles, while the y-axis represents the true quantiles. Darker shades indicate a higher number of test-set samples falling into the respective quantile categories, highlighting the distribution of prediction accuracy across the different methods. Numbers in the matrix represent the number of test-set samples in each bin.
4. DISCUSSION
4.1. Advantages of The FastImpute Pipeline
Traditional reference-based genotype imputation methods offer high accuracy but are computationally intensive and limited by reference panel accessibility. The size of the GRCh37 1000 Genome Project files can be prohibitive for many researchers, necessitating reliance on services like the Michigan Impute Server [3]. These services lack real-time genotype imputation capabilities and can cause delays in research and clinical decision-making due to server downtimes and long queue times. Additionally, these methods may underperform when target sets differ from discovery panels [25]. This could explain the variability in Beagle's R2 in our results, particularly at a few specific PRS313_BC SNPs. This variability could lead to inconsistent genetic risk assessments in underrepresented patient populations. Reference-free approaches [4, 5, 6, 7, 26] have been developed to address these issues. However, they are often too large for browser-based imputation and require computationally expensive retraining for imputing on different regions, limiting their practical clinical application.
This study introduces a lightweight, client-sided genotype imputation model that enhances both the accuracy and accessibility of polygenic risk score calculations through a serverless architecture - the first of its kind. A central pillar of this contribution is its open-source nature; the entire FastImpute ecosystem is publicly available (Application: https://aaronge-2020.github.io/FastImpute/, Code: https://github.com/aaronge-2020/FastImpute). This commitment to open-source ensures full transparency and reproducibility, enhances user privacy by processing all data locally on the user's device, and democratizes access to genetic risk assessment by removing computational and financial barriers. Our approach addresses key challenges in existing imputation methods by implementing a simple yet effective baseline linear regression model that balances performance, computational efficiency, and privacy. With significantly lower retraining costs compared to deep neural networks, the model's adaptability suits various genomic regions and pipelines while enabling real-time data processing on edge devices like smartphones, representing a significant advancement in point-of-care genetic testing and personalized medicine.
4.2. Clinical Significance
FastImpute offers a novel approach to genetic risk assessment with its efficient, client-side processing, demonstrating particular potential in the realm of direct-to-consumer (DTC) genetic data. The increasing popularity of DTC genetic testing has resulted in a wealth of readily available data. However, this data is often underutilized in terms of comprehensive risk assessment. While established clinical workflows often utilize targeted sequencing or other methods, FastImpute provides a valuable alternative for analyzing existing DTC array data, making it possible to extract further insights from this readily available resource. As demonstrated by our PRS313_BC case study for breast cancer risk assessment, FastImpute can empower individuals to explore their genetic predispositions using their DTC data and may facilitate more informed discussions about personalized risk with healthcare providers.
The baseline models trained using the FastImpute pipeline performed comparably in identifying high-risk individuals to more complex methods like Beagle when applied to 23andMe data. This is particularly relevant for breast cancer risk stratification, where Mavaddat et al. (2019) showed that women in the top 1% of the PRS313_BC distribution have a predicted risk approximately four times larger than those in the middle quintile, aligning with the UK NICE definition of high risk for breast cancer [15]. Our logistic regression model using phased data performed similarly to Beagle in identifying these high-risk individuals, with the linear regression model misclassifying only one additional patient (Fig. 7).
Moreover, FastImpute's ability to perform genetic risk assessments on edge devices such as smartphones enhances accessibility. Individuals can leverage their DTC data to gain insights into their risk profiles, potentially leading to more proactive discussions with healthcare providers. While not a replacement for comprehensive clinical evaluations, FastImpute serves as a valuable tool for individuals to better understand their genetic predispositions and seek further guidance if needed.
Furthermore, by processing data on edge devices, FastImpute addresses some of the computational and privacy barriers associated with traditional imputation methods. This approach could potentially extend genetic risk assessment to areas where computational resources and infrastructure are limited, or where individuals are hesitant to share their data with external servers.
While this on-device approach is a major step for individual access and privacy, bridging the gap to formal clinical utility requires overcoming several hurdles. There is a need for standardized guidelines to interpret PRS results and a clear path for integrating such tools into established clinical workflows. Critically, this process must involve genetic counselors to help patients navigate the probabilistic nature of PRS, ensuring the information is empowering rather than alarming. Therefore, we position FastImpute as a tool for risk exploration: one that enriches patient-provider discussions rather than serving as a standalone diagnostic instrument. Its current role as an exploratory tool highlights the need for extensive validation before it can be considered for clinical practice.
CONCLUSION
This study introduces FastImpute, a reference-free, lightweight genotype imputation model that enhances privacy and accessibility. By leveraging client-side deployment, FastImpute addresses the computational inefficiencies and privacy concerns of traditional methods and reference-free approaches.
Our PRS313_BC case study for breast cancer risk assessment shows that FastImpute can perform comparably to Beagle in identifying high-risk individuals, demonstrating its potential for real-time genetic risk assessments in clinical settings on devices like smartphones. This advancement could lead to earlier disease detection and more personalized treatments.
Future research could focus on expanding the training dataset to include more diverse genotyping chips and genomic regions, implementing superpopulation-specific calibration, and exploring more complex models to enhance imputation accuracy while maintaining the benefits of client-side deployment.
LIMITATIONS
While our study demonstrates the potential of FastImpute, we acknowledge certain limitations. First, our reconstruction of the 23andMe V5 panel relied on a sample of 119 users from openSNP.org [14]. Though this sample represents nearly half of the publicly available V5 data from 2020 onwards, larger, more representative datasets would enhance the generalizability of our findings. We were only trying to present a case study, so there is minimal impact on the validity of our results. Future collaborations with consumer genetic testing companies could provide access to more comprehensive data, allowing for a more robust analysis of various genotyping chips.
Second, our methodological choices were guided by our primary goal of creating a lightweight and portable pipeline. For instance, our data selection method focuses on regions within a +/- 500K base pair window with LD, following the default values provided by LDProxy. This window may overlook informative, long-range LD patterns and could potentially exclude some ancestry-specific information relevant for PRS accuracy. This choice was a deliberate compromise to maintain a small model size and ensure rapid, client-side processing. Future studies, however, could evaluate the impact of expanding this window to capture additional genetic variation, balancing imputation accuracy against computational cost.
Similarly, our study relies on linear and logistic regression models, which may not capture complex, non-linear genetic associations as effectively as deep learning methods could. This decision was guided by our feature selection strategy, which curated input SNPs based on strong linear correlation (R2), and by our primary goal of ensuring client-side deployability. While our preliminary tests showed that more complex models offered minimal performance gains for this feature set, they came at a prohibitive computational cost. Nonetheless, we acknowledge that this approach may not be optimal for all genomic contexts. Future work could explore hybrid models that incorporate non-linear effects while preserving a lightweight architecture.
Third, our proof-of-concept focused on a single polygenic risk score, PRS313_BC. Because this score is composed of 313 SNPs distributed across all autosomes, it served as a rigorous benchmark that inherently tests our workflow’s performance across a diverse set of genomic regions. However, the generalizability of our pipeline to polygenic risk scores developed for other diseases and traits, which may have different genetic architectures, has yet to be experimentally verified.
Finally, the PRS calculated in our web application is not calibrated based on the user's superpopulation. As we are only presenting an illustrative example, we believe this is beyond the scope of the current project. However, we recognize that this calibration is important, as the genetic distance between the user and our training dataset can influence the predictive power of our models. While out of scope for this initial proof-of-concept, future research should prioritize training superpopulation-specific models to enhance imputation accuracy and normalize PRS scores for clinical relevance.
AUTHORS’ CONTRIBUTIONS
The authors confirm their contribution to the paper as follows: A.G., J.B., P.K.: Study conception and design; A.G.: Data collection; A.G., J.B., X.W., P.K., J.S.A.: Analysis and interpretation of results; A.G., X.W., P.K., J.S.A.: Draft manuscript preparation. All authors read and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| AUC | = Area Under the Receiver Operating Characteristic Curve |
| DTC | = Direct-to-Consumer |
| IQS | = Imputation Quality Score |
| LD | = Linkage Disequilibrium |
| MAF | = Minor Allele Frequency |
| MHC | = Major Histocompatibility Complex |
| PRS | = Polygenic Risk Score |
| SNP | = Single Nucleotide Polymorphism |
| WGS | = Whole Genome Sequencing |
AVAILABILITY OF DATA AND MATERIALS
The data was available from: The FastImpute web application is freely accessible at https://aaronge-2020. github.io/FastImpute/.
The complete source code for the FastImpute pipeline has been deposited on GitHub and is available at https://github.com/aaronge-2020/FastImpute.
FUNDING
This work was funded by the National Cancer Institute (NCI) Intramural Research Program (DCEG/Episphere #10901).
ACKNOWLEDGEMENTS
Declared none.

