
Bioinformatic Analysis of a “Functional Cluster” Probably Related to Retinitis Pigmentosa
Abstract
Background:
Retinitis pigmentosa is an eye hereditary disease caused by photoreceptor death. One of the biggest problem is represented by its genetic heterogeneity, which has not yet allowed us to found all causative genes and how known ones could influence each other, leading to retinitis etiopathogenesis.
Objective:
To propose the possible relation between the “functional cluster” of vision dark adaptation, made of five phototransductional genes (RCVRN, GNB1, GNGT1, GRK7 and ARRB1), and retinitis pigmentosa onset.
Methods:
A bioinformatic approach was exploited: the starting point was searching through online database as PubMed and EMBASE to acquire information about the state of art of these gene. This step was followed by an in-silico analysis, performed by softwares as Cytoscape and Genecards Suite Plus, articulated in three phases: I) identification of common pathways and genes involved in; II) collection of previously detected genes; III) deep analysis of intersected genes and implication into etiopathogenesis of analzyed disease.
Results:
The whole in-silico analysis showed that all five gene products cooperate during phototransductional activation, expecially in the dark adaptation. Interestingly, the most exciting aspect regards the direct relation with several known retinitis pigmentosa causative genes, in form of protein interactions or other pathway correlations.
Conclusion:
Pathway analysis permitted us to hypothesize a possible role of analyzed genes in retinitis pigmentosa etiopathogenesis, also considering the key activity of their encoded proteins. Next step will be validating our hypotesis with functional assays to ensure the real meaning of this possible association, leading to new potential retinitis pigmentosa causative genes.
1. INTRODUCTION
Retinitis pigmentosa is a genetic disease involving the retina, the back portion of the eye, photosensitive and appointed to focus light signals towards the optical nerve first, then towards brain, after their transduction into the electrical stimuli [1]. It is an uncommon condition affecting about 1 in 4,000 people in the United States, and 1-5/10.000 in Italy [2], with an incidence just slightly lower than other rare genetic pathologies like Cerebral Cavernous Malformations (CCMs) [3, 4].
1.1. Biological Features of Retinitis Pigmentosa
The term “pigmentosa” deals with the characteristic appearance, during the advanced states of the disease, of abnormal areas of pigment into the retina. Degeneration involves both eyes and affects photoreceptors and Retinal Pigment Epithelium (RPE) [5], inducing a slow and progressive death in these cells and leading to lose ability to transmit brain the visual informations. Photoreceptors are particular light-sensitive cells: cones, so called for their characteristic shape, incorporate image and color details, and are mainly localized in the retina central part, called macula; rods, elongated and tapered, are involved in night vision and low light conditions, as well as in object motion perception, and are predominantly distributed in the peripheral zone [6]. Thanks to these receptors, for example, we can realize the danger that comes close to the corner of the eye, even if we are not able to perceive the details. Rods represent retinitis first and most involved photoreceptor system (cones are involved in advanced stages of disease, even if with a currently unknown mechanism): they undergo apoptosis, with a loss rate evidenced by ONL (Outer Nuclear layer, the retinal layer which contains them) decrease and retinal accumulation of pigment (rhodopsin) [7]. Generally, it can be assumed that the mere presence of the rods provides survival signals needed to keep “alive” cones.
1.2. Clinical Features of Retinitis Pigmentosa
About the clinical picture, the first symptom is (but not always) the decrease of crepuscular nocturnal visual acuity (difficulty in driving at night or move in dimly illuminated rooms), until you come to a narrowing of the peripheral visual field (difficulty in perceiving objects placed laterally or seeing the steps going down the stairs) and, in the final stage of the disease, the loss of central vision and blindness [8]. It can also be accompanied by cataract or deafness, events directly related to retinitis pigmentosa. Diagnosis [9] consists of fundus examination and visual field, followed by visus, electroretinogram and fluorescein angiography.
1.3. Genotypic – Phenotypic Features of Retinitis Pigmentosa
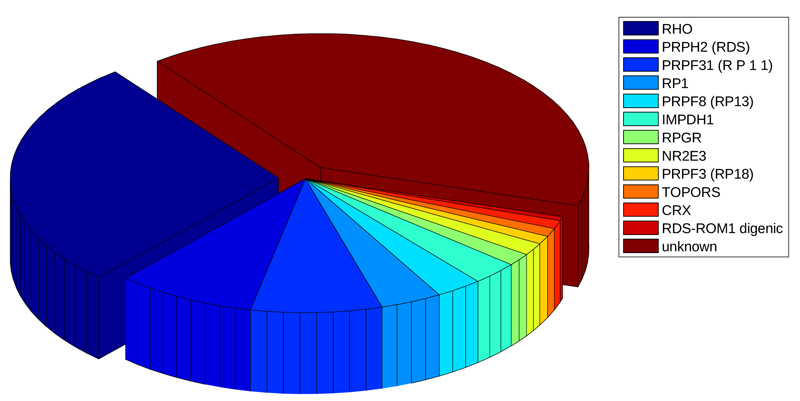
The disease progression rate and the age of symptom onset [10] vary according to many factors, including the pattern of genetic transmission. Today, it is known that a wide range of retinitis pigmentosa forms is caused by about 60 genes (Table 1), the most of them involved in phototransduction, canonical retinoid cycle in rods (twilight vision) and cones (daylight vision), inactivation, recovery and regulation of phototransduction cascade, cargo trafficking to the periciliary membrane and signal transduction [11]. A special mention goes to oxidative stress which, as for other hereditary pathologies [12], could determine the onset of retinitis pigmentosa [13].
| RETINITIS PIGMENTOSA | INHERITANCE | GENE | LOCALIZATION |
|---|---|---|---|
| 1 | Autosomal Recessive Autosomal Dominant |
RP1 | 8q11–q13 |
| 2 | Autosomal Recessive | RP2 | Xp11.23 |
| 4 | Autosomal Recessive Autosomal Dominant |
RHO | 3q21-q24 |
| 7 | Autosomal Dominant | ROM1, PRPH2 | 11q13, 6p21.1-cen |
| 9 | Autosomal Dominant | RP9 | 7p14.2 |
| 10 | Autosomal Dominant | IMPDH1 | 7q31.3-q32 |
| 11 | Autosomal Dominant | PRPF31 | 19q13.4 |
| 12 | Autosomal Recessive | CRB1 | 1q31-q32.1 |
| 13 | Autosomal Dominant | PRPF8 | 17p13.3 |
| 14 | Autosomal Recessive | TULP1 | 6p21.3 |
| 15 | X - Linked | RPGR | Xp11.4 |
| 17 | Autosomal Dominant | CA4 | 17q23 |
| 18 | Autosomal Dominant | PRPF3 | 1q21.2 |
| 19 | Autosomal Recessive | ABCA4 | 1p22.1 |
| 20 | Autosomal Recessive | RPE65 | 1p31 |
| 25 | Autosomal Recessive | EYS | 6q12 |
| 26 | Autosomal Recessive | CERKL | 2q31.2-q32.3 |
| 27 | Autosomal Dominant | NRL | 14q11.1-q11.2 |
| 28 | Autosomal Recessive | FAM161A | 2p15 |
| 30 | Autosomal Dominant | FSCN2 | 17q25 |
| 31 | Autosomal Dominant | TOPORS | 9p21 |
| 33 | Autosomal Dominant | SNRNP200 | 2q11.2 |
| 35 | Autosomal Recessive Autosomal Dominant |
SEMA4A | 1q22 |
| 36 | Autosomal Recessive | PRCD | 17q22 |
| 37 | Autosomal Recessive Autosomal Dominant |
NR2E3 | 15q23 |
| 38 | Autosomal Recessive | MERTK | 2q14.1 |
| 39 | Autosomal Recessive | USH2A | 1q41 |
| 40 | Autosomal Recessive | PDE6B | 4p16.3 |
| 41 | Autosomal Recessive | PROM1 | 4p15.3 |
| 42 | Autosomal Dominant | KLHL7 | 7p15.3 |
| 43 | Autosomal Recessive | PDE6A | 5q31.2-q34 |
| 44 | Autosomal Dominant | RGR | 10q23 |
| 45 | Autosomal Recessive | CNGB1 | 16q13 |
| 46 | Autosomal Recessive | IDH3B | 20p13 |
| 47 | Autosomal Recessive | SAG | 2q37.1 |
| 48 | Autosomal Dominant | GUCA1B | 6p21.1 |
| 50 | Autosomal Dominant | BEST1 | 11q13 |
| 51 | Autosomal Recessive | TTC8 | 14q32.1 |
| 54 | Autosomal Recessive | C2orf71 | 2p24.1-p23.1 |
| 56 | Autosomal Recessive | IMPG2 | 3q11.2 |
| 58 | Autosomal Recessive | ZNF513 | 2p24.1-p22.3 |
| 59 | Autosomal Recessive | DHDDS | 1p36.11 |
| 60 | Autosomal Dominant | PRPF6 | 20q13.33 |
| 62 | Autosomal Recessive | MAK | 6q22 |
| 64 | Autosomal Recessive | C8orf37 | 8922.1 |
| Bothnia Retinal Distrophy | Autosomal Recessive | RLBP1 | 15q26 |
| Juvenile Retinitis Pigmentosa, AIPL1-Related | Autosomal Recessive | AIPL1 | 17p13.2 |
| Leber congenital amaurosis 3 | Autosomal Recessive | SPATA7 | 14q31.3 |
| Leber congenital amaurosis 13 | Autosomal Recessive | RDH12 | 14q23.3 |
Mutations of these genes may be autosomal recessive (50-60%), dominant (30-40%) [14]; [15], X-linked (5-20%; the first gene linked to the RP phenotype was discovered twenty-six years ago on the X chromosome) [16] or sporadic (30%). Although these known genes cause the most of retinitis pigmentosa forms, there are many other ones that are still unidentified nowadays [11, 17-27], and several regulative aspects have to be discovered [28]. This scenario suggests that new genes could contribute to the genesis of pathology, and should be investigated in order to improve treatment for this disabling disease. Our approach is based on an in-silico analysis.
2. MATERIALS AND METHODS
The research was carried out looking for causative genes of eye – related pathologies in the following databases: PubMed, EMBASE and Web of Science. The following keywords were employed: retinitis pigmentosa, eye disease, cone - rode dystrophy, Leber's congenital amaurosis, in different combinations. Cross references were manually searched when needed. Due to space limits, not all references were quoted here; comprehensive reviews were quoted instead.
An in-silico analysis was, then, exploited to detect new genes related to retinitis pigmentosa, and it was performed in three different steps: I) identification of common pathways and genes involved in; II) collection of previously obtained genes; III) deep analysis of intersected genes and implication into etiopathogenesis of retinitis pigmentosa. In details:
- Identification of common pathways and genes involved in: in order to detect possible related functions and new related genes, an intersection of known causative genes and a research of common pathways were performed by Cytoscape software (http://www.cytoscape.org) and the three different plugins GeneMANIA (http://apps.cytoscape.org/apps/genemania), CytoKEGG (http://apps.cytoscape.org/apps/cytokegg) and BinGO (http://apps.cytoscape.org/apps/bingo).
- Collection of previously obtained genes: genes were collected from Kegg pathway database (www.kegg.jp), setting “Homo Sapiens” as organism and “phototransduction”, “retinitis”, “apoptosis”, “rod”, and “vision” as keywords.
- Deep analysis of selected genes and implication into etiopathogenesis of retinitis pigmentosa: all candidate genes have been added to “Pathway Analysis” query research of Reactome Pathway Database (www.reactome.org), BioGraph (www.biograph.be) and STRING (https://string-db.org), in order to establish relationship level between physiological and pathological functions, and between known causative genes. Finally, the job was completed with the help of Genecards Suite Plus, made of GeneAnalytics (https://ga.genecards.org/), PathCards (http://pathcards.genecards.org) and VarElect (http://varelect.genecards.org/), which provided a categorized list of matched tissues, cells, diseases, pathways, compounds and Gene Ontology (GO) terms to enhance gene set interpretation.
Moreover, in order to analyze common nodes related to each cellular process considered, a research of related phenotypes was performed in Malacards Human Disease Database (www.malacards.org).
3. RESULTS AND DISCUSSION
3.1. Cytoscape Analysis
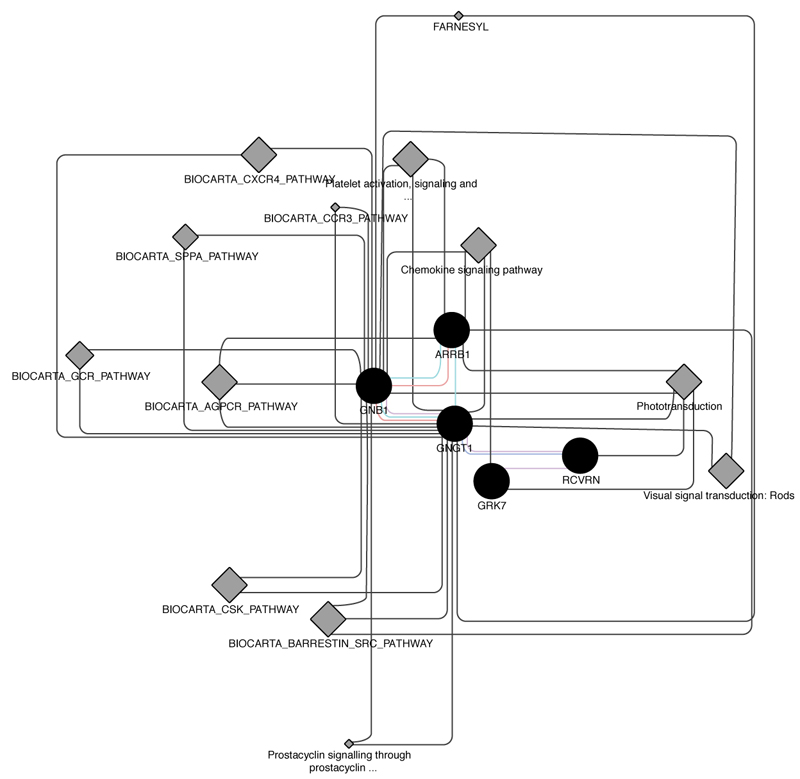
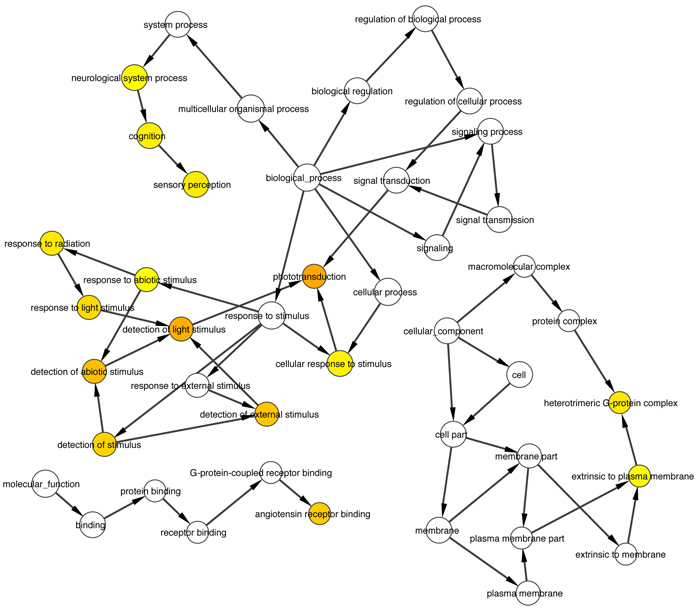
Intersection of about 50 causative genes for retinitis pigmentosa by Cytoscape and its plugins gave us about 30 proximal correlated genes, from witch we extracted 5 ones that are involved into phototransduction pathway, specifically in dark adaptation phases, which result the most altered one in pathological retinitis phenotype [29] (Figs. 1-3).
3.2. Phototransduction and the G-Protein Cascade [30]
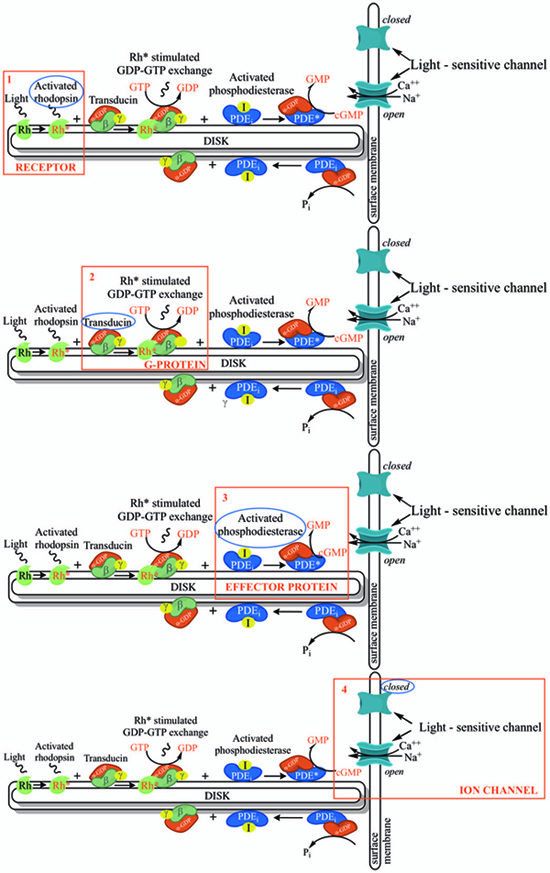
In order to understand at what level the filtered 5 genes could be involved in retinal function alterations, it is necessary to explain briefly the main phases of phototransduction. The transduction of light into a neural signal takes place in the outer segment of photoreceptors: in our study we underline what happens in rods, which are retinitis pigmentosa most involved cells. Rod outer segment shows a lipid bilayer membrane arranged as flattened “discs” (optical discs), which is strictly packed with the photopigment rhodopsin. Rhodopsin is a member of the superfamily of seven-helix, G-Protein–Coupled Receptor proteins (GPCRs). Differently from chemosensing GPCRs, rhodopsin presents its ligand, the light-absorbing “chromophore” retinaldehyde, prebound. The bound form, 11-cis retinal, acts as a strong antagonist, holding rhodopsin in its completely inactive state. Absorption of a photon of light isomerizes the chromophore to the the all-trans configuration, which rapidly triggers a conformational change in the protein, activating rhodopsin as an enzyme. The conformational change triggered by light causes rhodopsin absorption spectrum to shift into the UV, so that the pigment loses its visible color and is said to “bleach.” Long after the end of signaling, the Schiff-base bond that attaches the all-trans retinal is hydrolyzed, permitting the retinoid to dissociate from the apo-protein, opsin. In addition to rhodopsin (R), which is packed into the disc membrane at high density, three other classes of proteins cooperate into activation of the photoresponse: 1) an heterotrimeric G - protein (G), called transducin in rods, with a 1:10 stechiometry to rhodopsin; 2) the cyclic nucleotide phosphodiesterase (PDE), which hydrolyzes cyclic GMP (cGMP), the second cytoplasmic messenger, present at about 100 units per rhodopsin one; and (3) the Cyclic Nucleotide – Gated Channels (CNGCs) of the plasma membrane, which control the flow of electrical current into the outer segment. The first three steps involve proteins associated with the disc membrane (rhodopsin, which is integral, and the G-protein and PDE, which are connected by acyl groups), whereas the final two involve the cGMP, and its action at the plasma membrane (Fig. 4 summarize all five steps).

3.3. The Five Genes “Functional Cluster”: RCVRN, GNB1, GNGT1, GRK7 and ARRB1
RCVRN: this gene encodes a member of the recoverin family of neuronal calcium sensors, which contains three calcium-binding EF-hand domains. It may delay phototransduction cascade termination in the retina by blocking the phosphorylation of photo-activated rhodopsin [31, 32]. Recoverin may be the antigen responsible for cancer-associated retinopathy [33].
GNB1 and GNGT1: heterotrimeric guanine nucleotide-binding proteins (G proteins), which transduce extracellular signals received by transmembrane receptors to effector proteins [34]. They are membrane bound GTPases that are linked to 7-TM receptors. Each G protein contains an α-, β- and γ-subunit and is bound to GDP in the “off” state. Ligand-receptor binding results in detachment of the G protein, switching it to an “on” state and permitting Gα activation of second messenger signalling cascades. Transducin is one of these proteins, found specifically in rod outer segments, where it mediates the activation of a cyclic GTP-specific guanosine monophosphate phosphodiesterase by rhodopsin [35]. GNB1 encodes a β subunit, while GNGT1 the gamma one [36, 37]. The α subunit is encoded by GNAT1 gene. The β and γ chains are required for the GTPase activity, for replacement of GDP by GTP, and for G protein-effector interaction (provided by RefSeq, Sep 2013).
GRK7: This gene encodes a member of the guanine nucleotide-binding protein (G protein)-coupled receptor kinase subfamily of the Ser/Thr protein kinase family (GRK) [38]. It is a retina-specific kinase involved in the shutoff of the photoresponse and adaptation to changing light conditions via cone opsin phosphorylation, including rhodopsin (RHO) [39]. It is believed that the movements of intracytoplasmic loop of activated RHO are responsible for the activation of the GRK7, in a way similar to GRK1 [40] (Fig. 5)).
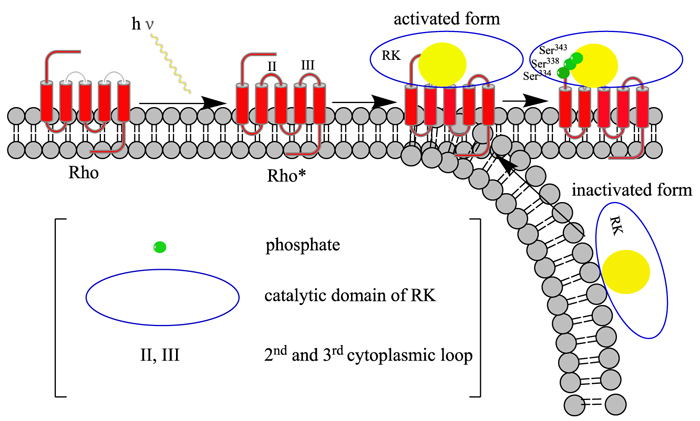
ARRB1: Members of arrestin/beta-arrestin protein family are thought to participate in agonist-mediated desensitization of G-protein-coupled receptors and cause specific dampening of cellular responses to stimuli such as hormones, neurotransmitters, or sensory signals [41]. Arrestin beta 1 is a cytosolic protein and acts as a cofactor in the beta-adrenergic receptor kinase (BARK) mediated desensitization of beta-adrenergic receptors. During homologous desensitization, beta-arrestins bind to the GPRK-phosphorylated receptor and sterically preclude its coupling to the cognate G – protein (Fig. 6); the binding appears to require additional receptor determinants exposed only in the active receptor conformation [42]. The beta-arrestins target many receptors for internalization by acting as endocytic adapters (CLASPs, clathrin-associated sorting proteins) and recruiting the GPRCs to the adapter protein 2 complex 2 (AP-2) in clathrin-coated pits (CCPs). However, the extent of beta-arrestin involvement appears to vary significantly depending on the receptor, agonist and cell type. Internalized arrestin-receptor complexes traffic to intracellular endosomes, where they remain uncoupled from G-proteins [43]. Beta-arrestins function as multivalent adapter proteins that can act as signaling scaffold for MAPK pathways, like several other proteins as CCM proteins [44, 45], in the so-called beta-arrestin signalosomes [46]. Moreover, they can be involved in IGF1-stimulated AKT1 signaling leading to increased protection from apoptosis, in activation of the p38 MAPK signaling pathway and in actin bundle formation, in F2RL1-mediated cytoskeletal rearrangement and chemotaxis, and in AGTR1-mediated stress fiber formation by acting together with GNAQ to activate RHOA [47].
3.4. Common Pathways and Possible Involvment in Retinitis Pigmentosa
3.4.1. Reactome Analysis
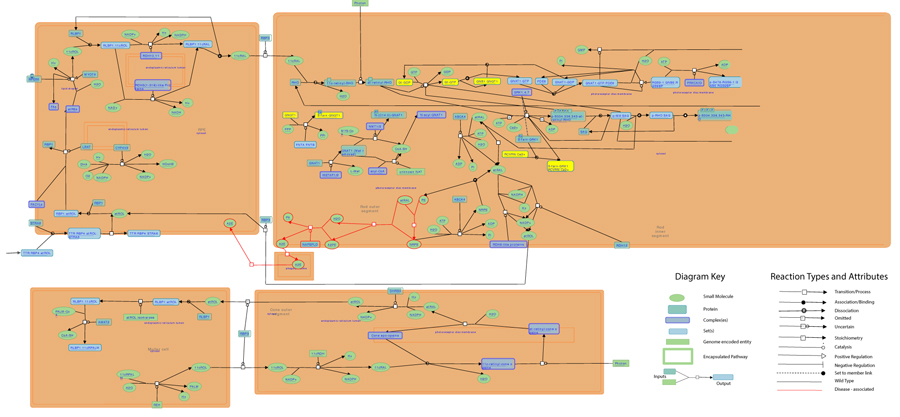
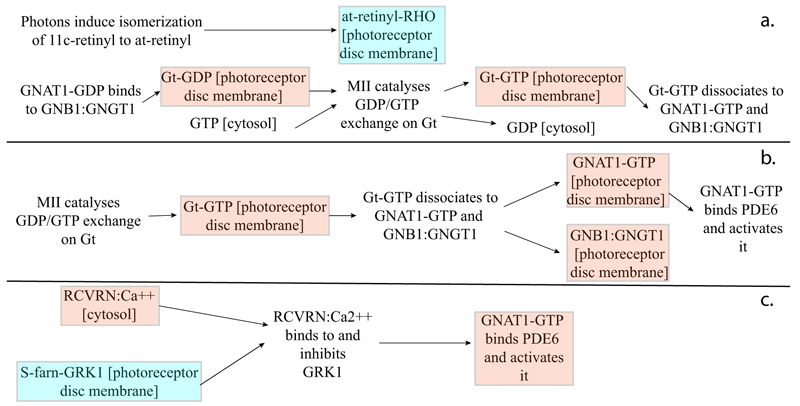
As highlighted by the pathway database Reactome, all five gene products cooperate during phototransductional activation (Fig. 7), expecially in the dark adaptation, as confirmed by KEGG (Fig. 8). In darkness, the G protein transducin (Gt) is attached to the disk membrane surface with a GDP bound to it and it is inactive. Photoactivated rhodopsin (R*) catalyzes the exchange of GTP (present in a higher concentration than GDP) for GDP bound to Gt. Upon GTP/GDP exchange, Gt is released from R* and the Gtα with GTP bound (GNAT1 GTP) dissociates from Gtβ-Gtγ subunits (GNB1:GNGT1). This mechanism was deciphered from bovine experiments [48]. R* proceeds to activate additional Gt molecules, making this reaction the first amplification step in the phototransduction cascade. A single activated rhodopsin molecule activates tens of Gt molecules. R* must be deactivated to terminate the single photon response, and it happens after R* binds a rhodopsin kinase family member (as GRK7), a serine/threonine protein kinase [49]. GRK is activated by R* whereupon it phosphorylates R* at multiple serine and threonine sites (six in total) on its C terminus. Increasing phosphorylation progressively reduces the rate at which R* can activate transducin, but complete deactivation occurs only after arrestin (ARRB1) binds to and sterically caps R* [50]. A substantial fraction of GRK is bound to recoverin (RCVRN) in darkness, when internal Ca2+ levels are high (Fig. 9).
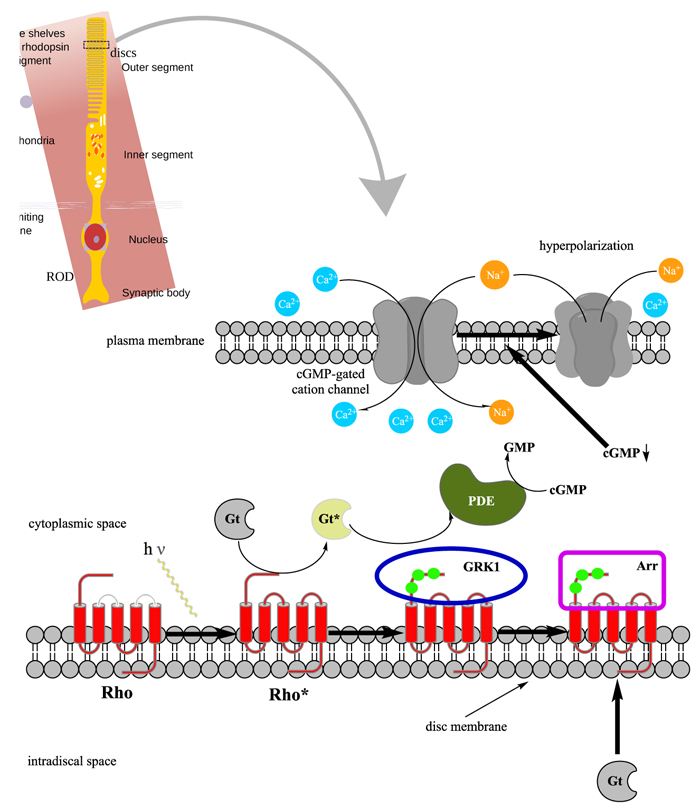
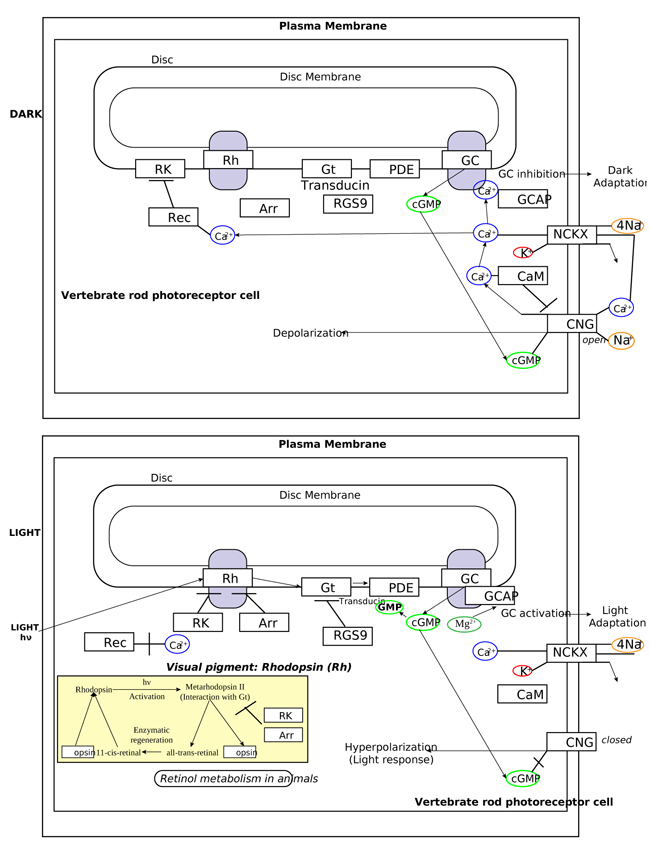
RCVRN is an EF-hand protein [50] that functions as a myristoyl switch. With Ca2+ bound, the myristoyl group is exposed to attach RCVRN to the membrane. When Ca2+ levels drop with light exposure, Ca2+ dissociates from RCVRN and GRK is released. Higher levels of free GRK1 accelerate the phosphorylation and shutoff of photoexcited rhodopsin (R*). However, this feedback mechanism proceeds too slowly to impact the single photon response that was responsible for causing the fall in Ca2+. Instead, it operates during light adaptation, where the light-induced fall in Ca2+ primes the rod to release GRK1 to act after subsequent photoisomerizations of rhodopsin. RCVRN also serves as a Ca2+ buffer within the rod outer segment.
3.4.2. BioGraph Analysis
Thanks to BioGraph web service, we could conjecture how these genes should be involved into etiopathogenesis of retinitis pigmentosa. All five analyzed genes share annotation or pathways with already known causative ones: in detail, they are TULP1 (required for normal photoreceptor function and for long-term survival of photoreceptor cells, interacts with cytoskeleton proteins and may play a role in protein transport of these cells), PROM1 (during early retinal development acts as a key regulator of disk morphogenesis; it is also involved in regulation of MAPK and Akt signaling pathways), RP1 (encoding a MAP needed for the correct stacking of disc in the outer segment of photoreceptor), RPGR (guanine-nucleotide releasing factor, which plays a role in ciliogenesis and in intraflagellar transport processes by regulating actin stress filaments and cell contractility), CNGA1 (the encoded protein can be activated by cyclic GMP which leads to an opening of the cation channel and thereby causing a depolarization of rod photoreceptors), GUCA1B (calcium-binding protein that activates photoreceptor guanylate cyclases; this Ca2+-sensitive regulation of GC is a key event in recovery of the dark state of rod photoreceptors following light exposure), PDE6A-B (these genes encode alfa and beta subunits of the phosphodiesterase 6 holoenzyme, which regulate the rod cGMP concentration, an important regulator of cell membrane current), and CNGB1 (subunit of cyclic nucleotide-gated channels, nonselective cation channels, involved in the regulation of ion flow into the rod photoreceptor outer segment (ROS) in response to light-induced alteration of cGMP intracellular levels). GNGT1 shares many targets of GNB1, but also shows a protein interaction with RHO and involvement in eye photoreceptor cell development and light signal transduction, annotating with RGR (putative retinal G-protein coupled receptor for all-trans- and 11-cis-retinal; it binds preferentially to the former and may catalyze the isomerization of the chromophore by a retinochrome-like mechanism). RCVRN stands for the calcium sensitive guanylate cyclase activator activity, requested for its fine molecular regulation. GRK7 evidenced a protein interaction with PDE6D (another subunity of the PDE complex, that also binds to prenyl groups of proteins to target them to subcellular organelles called cilia) and annotation with genes appointed to vision, FAM161A (involved in ciliogenesis) and SPATA7 (required for the stable assembly and localization of the ciliary RPGRIP1 protein complex in the connecting cilium). ARRB1, instead, shows a fundamental protein interaction with SAG (member of arrestin/beta-arrestin protein family, is a major soluble photoreceptor protein that is involved in desensitization of the photoactivated transduction cascade) and DPY30, component of a multiprotein histone methyltransferase complex, which regulates the causative PRPF3 gene (participates in pre-mRNA splicing). Summarizing, the five clustered genes are mainly involved in phototransduction, eye photoreceptor cell development and calcium sensitive guanylate cyclase activator activity pathways, medianting interactions with light signal transduction key proteins. Alterations in these processes could lead to retinitis pigmentosa (Fig. 10).
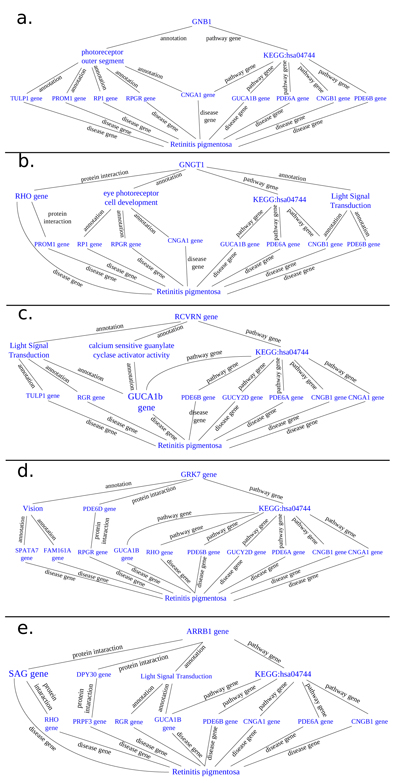
3.4.3. STRING Analysis
All these cooperative genes are also related on String phototransductional pathway graphical scheme (Fig. 11), which highlights how they give rise to “functional clusters”, units that support the same biological activity. The five analyzed genes result are mainly involved in G – protein signaling modulation, as evidenced by interaction with G – protein subunits and signaling regulators families, together with phosphoducin – like protein (PDCL), G – protein signaling modulator 1 (GPSM1) and adrenoceptor beta 2 (ADRB2). Moreover, the connection between the considered five gene cluster and cyclic nucleotide gated channel family seems to be very interesting, as proven by interactions with cyclic nucleotide gated channel family proteins. It is evident that all considered genes, directly or not, share the final target RHO, which presents the highest frequency of autosomal dominant retinitis pigmentosa known mutations found until today (Fig. 12).

3.4.4. GeneAnalytics and VarElect Analyses
To further substantiate the possible relation between examined genes and retinitis pigmentosa, we used the Genecards Plus Suite, made of several modules. The Gene Analytics gave us positive scores about tissue (eye = 0.83) and cells (retinal cells = 1.35 and mature rod cells = 0.91) where their encoded proteins are expressed. Based on the function, other useful scores came from gene ontology biological processes (phototransduction = 25.14, regulation of rhodopsin mediated signal pathway = 24.71, signal transduction = 19.41) and from superpathways filtered by Reactome itself, Qiagen, GeneGo, BioSystems, KEGG and PharmGKB (the phototransduction cascade = 43.08 and diseases associated with visual transduction = 19.97). The last evidence from Gene Analytics regards compounds or small molecules related to query gene set, in order to deliver possible therapeutic approaches against certain pathology: in our case, farnesyl (19.03), guanosine monophosphate (10.33), guanosine diphosphate (9.60) and metarhodopsin II (9.44) have the best scores. The other main module of the suite is VarElect, which associates a phenotype to a list of candidate genes. The five query genes were correlated with retinitis pigmentosa phenotype, and results were encouraging: three of them (GNB1, GNGT1 and RCVRN) were already objects of study in query disease researches, while the other two (GRK7 and ARRB1) have never been associated until now.
CONCLUSION
In this work, we tried to relate a “functional cluster” made of five genes (GNB1, GNGT1, GRK7, RCVRN and ARRB1) with retinitis pigmentosa pathology. We chose this group of genes due to their correlation with dark light adaptation, the most severe consequence of query disease. Pathway analysis, with the help of many qualified online databases, permitted us to hypothesize their possible role into etiopathogenesis of retinitis pigmentosa, also considering the key activity of their encoded proteins (rhodopsin kinase and transducin with own inhibitors). Next step will be validating our hypothesis with functional assays to ensure the real meaning of this possible association, with the final purpose of discovering new retinitis pigmentosa causative genes. Such proposal will be very useful to develop new personalized diagnostic approaches, followed by new therapeutic ones.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICTS OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

