
Identification of Protease Inhibition Mechanism by Iturin A against Agriculture Cutworm (Spodoptera litura) by Homology Modeling and Molecular Dynamics
Authors Info & Affiliations
Abstract
Objective:
Spodoptera litura, otherwise known as cutworm, belongs to the Noctuidae tribe, which is a severe scourge for numerous crop systems and is considered one of Asian tropical agriculture's most important insects. The world's leading environmental threats are plant pests, and the already commercialized pesticides are extremely poisonous and non-biodegradable and maybe additional residues harmful to the ecosystem. The increased resistance in pests often demands the need for advanced, active pesticides that are environmentally friendly and biodegradable.
Methods:
In the current work, the significance of proteases for the Spodoptera litura digestive system has been determined by the use of microbial metabolite protease inhibitor (Iturin A) in silico models. In the present study, we developed a model based on sequence structural alignment of known crystal structure 2D1I protease from Homo sapiens. The model's reliability evaluation was performed using programs such as PROCHECK, WHAT IF, PROSA, Validate 3D, ERRAT, etc.
Results:
In an attempt to find new inhibitors for Protease docking, the study was carried out with Iturin A. PMDB ID for the produced protease model was submitted to identify new inhibitors for Protease docking, and its accession number is PM0082285. The detailed study of enzyme-inhibitor interactions identified similar core residues; GLU215, LEU216, LYS217, and GLU237 have demonstrated their role in the binding efficacy of ligands.
Conclusion:
The latest homology modeling and docking experiments on the protease model will provide useful insight knowledge for the logical approach of constructing a wide spectrum of novel insecticide against Spodoptera.
1. INTRODUCTION
Spodoptera litura's larval stage is very risky for many crops, resulting in hosts that can induce defoliation [1]. This pest belongs to the noctuid group of parasites that has critical foliar feeders causing severe injury to tubers and roots [2]. The larval stage of S. litura acts as a cutworm when eating host leaves [3]. It causes the host plant to be stunted, resulting in late development of fruits. This dangerous pest S. litura also causes leaf skeletonization [4]. The leaves remain together during the early developmental stages when colonial mass extrudes out of the embryos. The adult larval phases are, however, solitary. They trigger several small feeding points on the vine, spreading later on to the entire leaf [5]. Several holes can appear and spread all over the sheet as a result of this harmful feeding. The holes may show different sections of the host, such as young stalks, bolls, and buds.
In certain instances, the larva destroys the plants by hitting the tips of the shoot and lowering them. The compartmentalization of proteases in specialized regions in the S. litura enteric guts could lead to better stability and improve action against plant-based protease inhibitors [6]. Nonetheless, several theories have proposed that protease inhibitors can work by attenuating metabolic process enzymes such as proteolysis, which is helpful in nutrient assimilation [7, 8]. Due to evolutionary resistance mechanisms, these pests have begun to develop a new class of gut proteases that may not be inhibited by such conventional protease inhibitors [9, 10].
The usage of synthetic pesticides is associated with both activating environmentally harmful agents, as well as strengthening the process of tolerance in specific pesticides [11]. There is, therefore, an urgent need for the production of healthy, complementary, and novel biopesticides derived from microbes [12]. The Bacillus group is the storehouse and capable of creating a new form of pesticide metabolite. The hot topic in demand is the production of the Bacillus genus of biopesticidal lipopeptides, namely Surfactin, Iturin, and Fengycin. The lipopeptide family biomolecule Iturin A is the most prevalent metabolite for pathways of controlling [13]. This biomolecule consists of heptapeptide with amino fatty acid and has extreme pesticide activity. Iturin A allegedly offers a broad range of antifungal actions. Iturin A has also shown to be an important insecticide against malarial insects and was popular. Due to the overload of these residues in the environment, the entire industrial sector is looking for safe, novel, bioactive pesticides that are safe for animals and ecofriendly biopesticides tools [14]. This has provided the researchers with a new direction to think about how Iturin A can be used as an outstanding biocontrol agent that is effective, biodegradable, and can substitute conventionally dangerous chemical pesticides [15].
2. MATERIALS AND METHODS
The calculations in the present analysis were performed using AMD Opteron Quad-core 2.10 GHz and 8 GB RAM. Protein tasks were designed using Modeler 9v7. Besides, molecular simulations were evaluated using Gromacs 4.6; and docking experiments were performed using edition GOLD 3.0.1. Unless specified, default settings were used for all calculations and tests.
2.1. Sequence Alignments
The amino acid sequence of Spodoptera enzyme protease was retrieved from UNIPROT in FASTA format [16]. The possible practical models for the protease series were obtained by conducting Blast P (search against Protein Data Bank entries for all proteins) [17]. Blast algorithm senses the resemblance of embedded models in global orientation, and sequences of homologs are matched with the Clustal-X tool [18]. The method estimates the best sequences, depending on the chosen sequences' similarities and differences using Modeller 9v7 Tools, a workstation for silicon graphics [19]; the selected protease sequences were used for comparative modeling. The software produces a tertiary protein structure with satisfactory spatial constraints enforced by the prototype structure sequence alignment [20]. Using regular parameter sets and repositories, the software was studied. The 3D protein model was acquired by increasing the capacity of atomic likelihood thickness while at the same time, restricting the abuse of data controls. The progression approach provides a variety of homology models for conducting appropriate conformational analysis of increasing complex site buildup. Fundamental methods are seen to offer an appropriate analysis by a number somewhere in the range of 10 and 100 models [21]. All acquired models are subjected to a re-enacted reinforcement convention that can be reached at Modeler to streamline the tight cooperation. The Protease's generated 3D framework was developed using the SPDBV code, which is an extensible package for sub-atomic perception tools used to produce precise details and appealing nuclear knowledge representation. Of the 100 types, the best one with the lowest root means square deviation (RMSD) confidence was chosen for further analysis while superimposed on form 2D1I.
2.2. Molecular Dynamics (MD) Simulations
The simulations on Molecular Dynamics (MD) were performed to verify the selected sequence alignment and eliminate the non-relevant derivative from the homology framework. The model selected for the protease was subjected to the reconstruction of subatomic elements utilizing the Gromacs 3.2.1 package and precisely the power field of 43A1 (Gromacs 96) [22]. The best Protease model was placed with an SPC water model in a truncated octahedron shell, and particles (Na+ and Cl-) were assembled. To remove near interactions with Van der Waals, both hydrogen ions, dust, and water atoms were subjected to 50 rounds of vitality minimization using the steepest vitality curve. The system was then sent for a period of 1ps to a brief MD replication with location controls and eventually subjected to complete MD replay. Protease model was introduced at 300k to 5000 ps, with no restrictions, using two fs of joining time. The conveying speeds were reassigned by Maxwell-Boltzmann at each progression. Lincs algorithm was related to all connections in a hydrogen molecule with a tolerance of 10-5 A°. After the equilibration stage, no additional controls were added. The leisure mixture was broken down concerning future longevity, the original RMSD model framework, and root-mean-square variation The investigation was determined concerning the c alpha spine structures, and each of them, which facilitates the outline from the directions, was superimposed on the initial compliance to evacuate any impact of general translation and revolution [23-26].
2.3. Assessment of the Built Model
In this step, the energy was minimized, its stereochemical parameters of in protease model with geometric quality of residue interaction efficiency, backbone conformation, the energy profile and the residue contact of the structure were detected by What if, Prosa, Procheck, verify 3D and Errat [27-31]. The final variant was chosen based on the high scoring of all built-in protease function assessment measures. Pdbsum created the protease model's secondary structures, and the Motif scan server used to classify different domains in the constructed model.
2.4. Docking Analysis
After the final protease model was chosen, docking tests were conducted utilizing the GOLD 3.0.1 software. There is so far no bacterial metabolite that could inhibit protease activity in S. litura. To identify a new protease enzyme inhibitor in the current research, Iturin A has been screened for the potential to obey the properties of Lipinski's five law, namely Mw (molecular weight), Log p (partition coefficient), TPSA, H-bond donors, and H-bond acceptors. The default protocol software GOLD 3.0.1 was used for forecasting the ligand's interactions with biomolecular targets [32]. Partial unified atomic charges and atomic solvation parameters were allocated to the protease molecule. Iturin A drawn in the 2-Dimensional format was translated using the Chemsketch 12.0 program to the 3-Dimensional template. The adding hydrogen atoms occupied all the negative valences in the ligand molecule. Additionally, all-atom forms were tested before docking in each ligand. The auto grid module allowed setting a grid map with x, y, z parameters for each ligand molecule (default 60 x 60 x 60 x 60). The grid box protected the protease enzyme docking site by providing room for ligand (translational and rotational) movement. The grid points are located at 0.375A0. The docking program was set to 2,000,000 and 27,000 and placed randomly as 300 initial populations, pressure, and number of operations, respectively. The identified convergence rate, mutation rate, and elitism were 0.8, 0.02, and 1.0, respectively. Local search parameters were set for the analysis of scalable docking genetic algorithms with a sample size of 300 individuals at 100 separate runs. After the docking studies had been completed, the Iturin A interactions with protease were described in the form of lowest docked energy, cluster RMSD values, and free binding energy. Using python molecular display, the docking tests were analyzed. It provided a clearer description of the amino acids involved in ligand binding in protease [33].
3. RESULTS AND DISCUSSION
Protease from Spodoptera litura displayed 365 amino acids and the sequence was retrieved from UNIPROT in Accession No J9XNW6 carrying FASTA format. The crystal structure was downloaded from Protein Data Bank (PDP), such as 2D1I showing gene homology (47 percent) with Protease series. The sequence identification of selected templates as 47% was observed with a small estimation of 30% structure personality to establish a pattern of homology. The PDB crystal structure with Id 2D1I suggesting homology grouping with protease arrangement was modified using separate Clustal-X succession arrangement apparatus (Fig. 1). Compared to sorting, there are not many differences and anomalies in the distribution of successions at the framework loops [34]. Inboxes have been seen aligning the protease pattern with models showing 12 insertions and 154 retained residues.

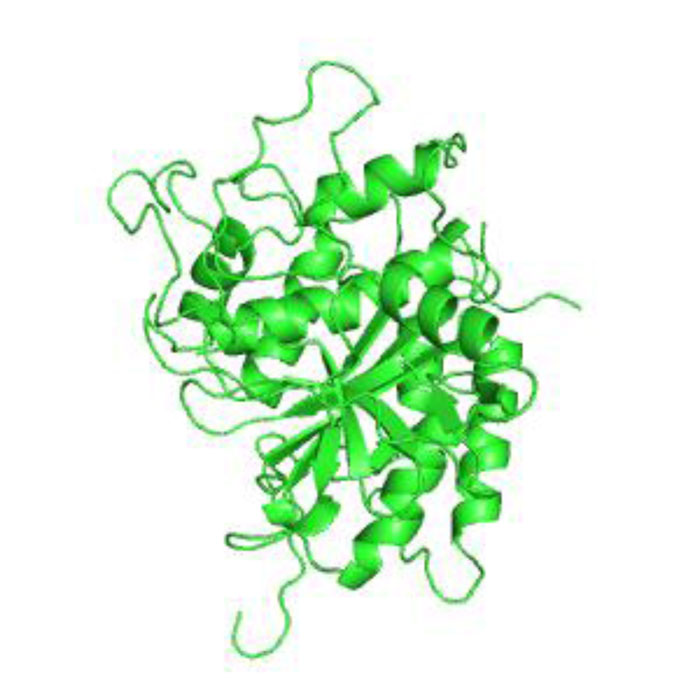
The 3-D protease model was constructed utilizing Homosapiens' crystal structure instructions (PDB Id: 2D1I 1.05A °) centered on the arrangement (Fig. 2). The protease homology approach was achieved by using the software Modeler 9v7. The prototype (2D1I) was selected from among the 100 models for further studies focused on the minimal Root Mean Square Deviation (RMSD). The RMSD spine superposition mirror value of the tertiary protease (Protease-2D1I) structure was observed at 0.61 A (Fig. 3). The spine superposition mirror and critical protection of the complex with low RMSD values are challenging for the homology modeling design framework [35].
3.1. MD Simulation Analysis
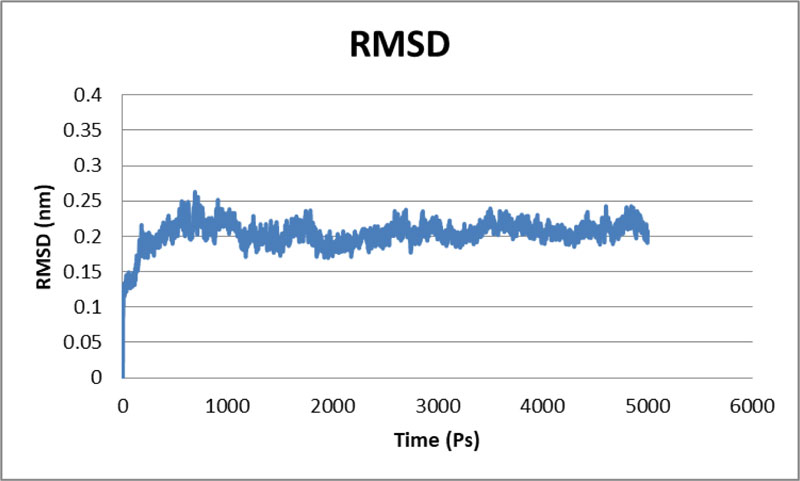
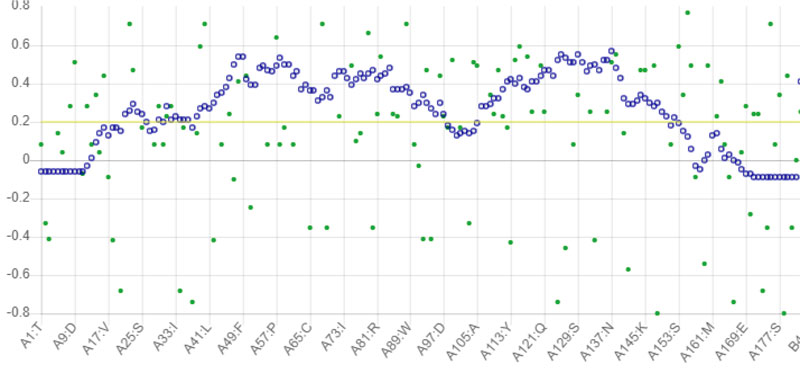
The stability of the proposed Protease model was tested using MD calculations. For the whole production phase of the 5000ps, MD reenactment runs in the instructions were clear. An elevation in the RMSD esteems in the primary 1000ps of replication was observed for protease, and in the ensuing reenactment, period achieved the stability. In the key 1,000 ps, an increase in the reward was inferable from the protein's relaxing activity or the strength reported was inaccurate. The standard protease RMSD was observed to be-0.422308 nm when measured from 5000 ps (Fig. 4). The map showed more important RMSF values for the deposits at the N-terminal locale. Most severe variances were found-0.2 nm for total protein. Large shifts in the spine are expected to arise on district adjusted, while places with small RMSF contribute mainly to the unbending beta-alpha-beta overlap. The poor valuation of RMSF worth in the RMSF specification shows the inaccurately arranged regions of room terminals. Not several vacillations reached 0.6 nm, and still fewer variances reached 0.8 nm for the whole protein. The Protease model's RMSF demonstrated that there were fewer changes in all dynamic site deposits, which assume a significant job in ligand binding [36].
3.2. Validation of the Protease Model
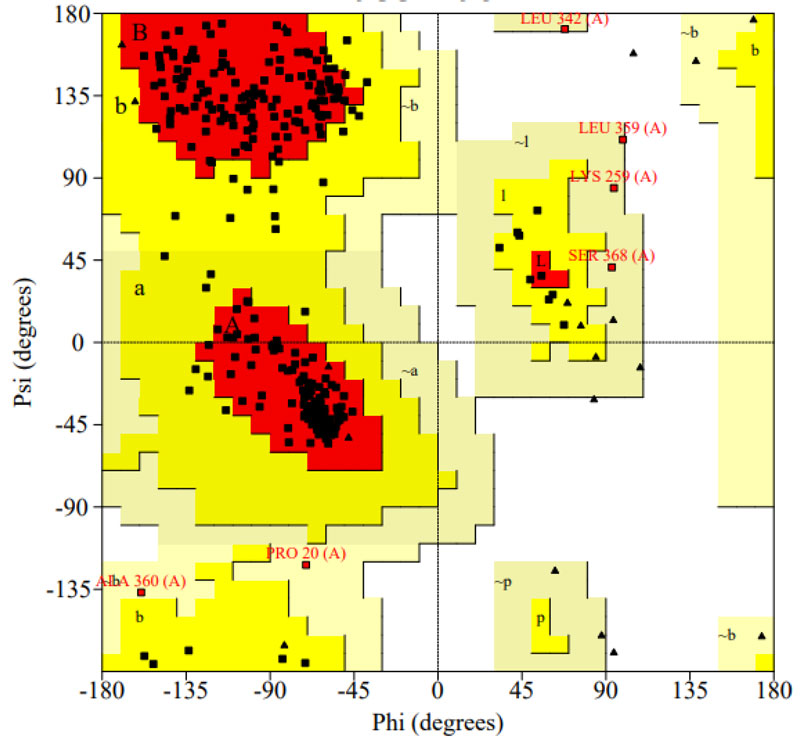
The selected programs such as PROCHECK, PROSA, VERIFY 3D, and ERRAT were used to carry out stereochemical constraints and Protease model structural evaluation. Ramachandran plot modeling for the protease model demonstrated the correct contribution of all amino acids in the phi and psi regions. The analysis shows that 94.0 percent of protease structure residues in the most desirable zone, 4.2 percent of residues in the additionally permissible field, 1.8 percent in reasonably allowed regions, and agree with the blueprint (Fig. 5) [37]. The consistency of the designed model suggested that 90 percent of contaminants were geometrically appropriate at the permissible area. The Protease model's main chain parameter was performed with PROCHECK, which revealed that the percent residues and omega angle, Bad contacts\100 residues, Zeta angle, H-bond strength, total G-factor values (Table 1) are observed within the permitted area. The protease model's side chain parameter shows the Trans standard deviation, Gauche plus standard deviation, Chi-gauche minus standard deviation, Chi-2 standard deviation and, chi pooled standard deviation values are in perfect alignment with anticipated benefits [38-40].
| Number of residues in the favoured region: | 315 (94.0%) |
| Number of residues in the allowed region: | 14 (4.2%) |
| Number of residues in the outlier region: | 6 (1.8%) |
The intensity of the contact was tested using PROSA. This displaces energy profiles and Z-scores (quality overall). Z-score on the protease is -5.24, within a range of the Z-score 2D1I as 4.5. Complete residue energy has contributed to pair energy, combined energy, and surface energy, and all are shown against models except in the starting area. The study has shown that the protease's average protein packing consistency is -0.843, backbone conformation -0.364. The RMS Z-scores for bond angles are bond length: 1.154, bond angle: 1.647, all within native template structure range.
The protease model's G- factor was shown as -0.2; the full 0 to –0.5 ranges as appropriate. ERRAT observed correlations between consistency and bonding (High quality and high score). The normal size for this high-quality configuration is > 50. In this analysis, the Errat value was observed at 92.346 for Protease and 94.24 for template 2D1I (Fig. 6). The protease was also tested by validated 3D — the compatibility score above zero in the protease 3D graph being reviewed corresponded to the friendly side-chain setting. The Protease model's secondary structure study was performed via the online database Pdb-sum, which offers details regarding helices, chains, threads, turns, etc. The present model consists of 12 helices, 14 Beta strands, and 8 sheets (Fig. 7). Proteins have a secondary structure that is easily recognizable, including unique categories and roles. Areas are individual, with unique roles that relate to the whole family functioning. The protease model was introduced to the scan server for the motif. It provided one region (21-302), which is the substratum-binding domain. It plays an important part in connecting a nucleotide to sugar.
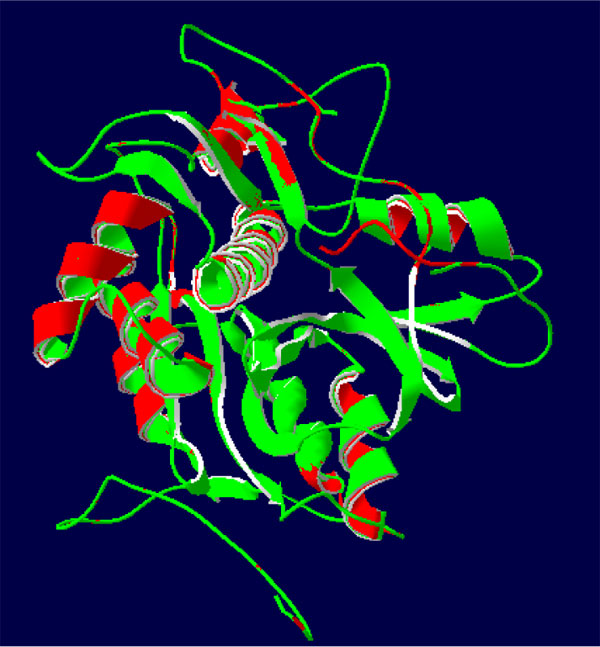
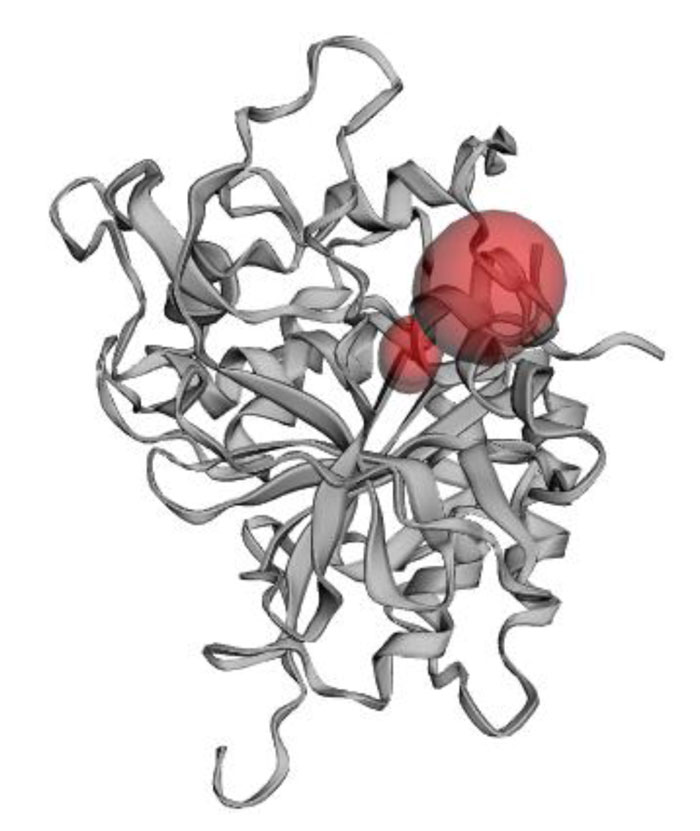
3.3. Docking Studies
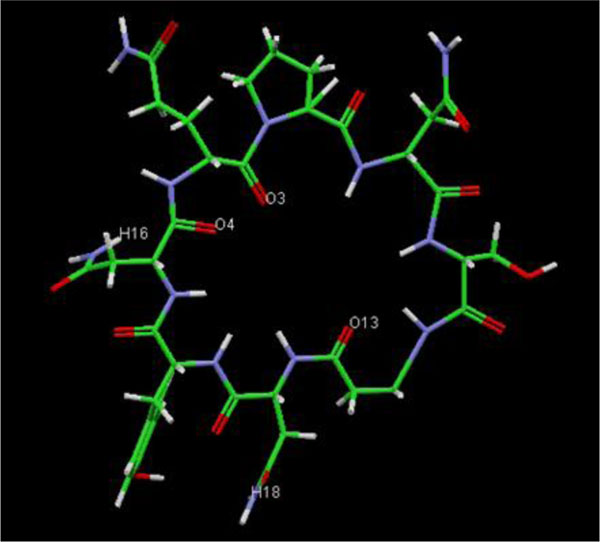
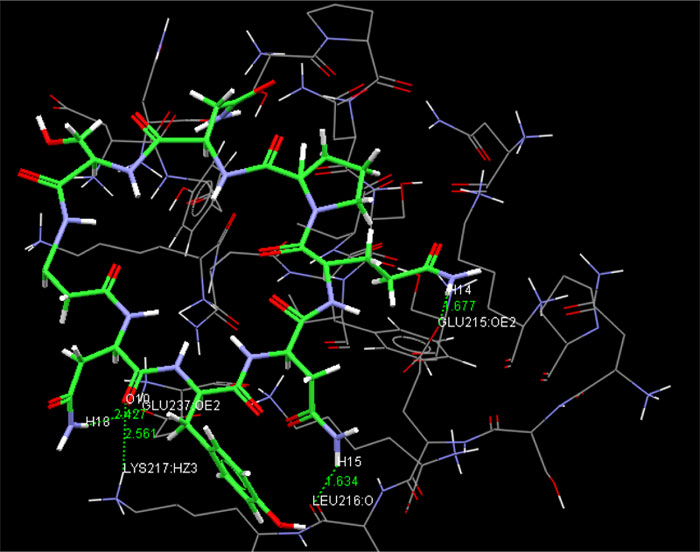
The present research showed that the pharmacophore models were generated from Iturin A structural manipulations. Iturin A experiments were performed with Spodoptera litura protease, a digestive enzyme. The compound (Iturin A) structures shown in (Fig. 8) were developed and configured with chem sketch tools. The measurements for the docking were performed using GOLD 3.0.1. A library of 10 iturin A confirmation was developed and screened to meet minimum ADME requirements for further study using Molinspiration [39, 41, 42]. The fulfilling rule of violations five with zero in the protease model was found among the 10, the top-ranking first 3 lead confirmations. Iturin A's final docking file provided an active amino acid site within the catalytic pocket (Table 2). The study was verified using free binding energy, lowest docked energy, and determined RMSD values. Iturin A showed the strongest associations with the active protease amino acids, namely GLU215, LEU216, LYS217, and GLU237 (Fig. 9). The whole clusters of Iturin A docking confirmations display destructive, linking energies. Of all Iturin A docking conformations, the best expected binding energy of -16.52k.cal/mol and RMSD of 0.24Å were indicated for the protease model (Table 3). Docking findings of each Iturin A with Protease were evaluated using python molecular viewer.
| Fitness | S(hb_ext) | S(vdw_ext) | S(hb_int) | S(int) | Ligand name | ||||
| -16.52 | 9.07 | 24.81 | 0.00 | 59.72 | Iturin A | ||||
| S(hb_ext) S(vdw_ext) S(hb_int) S(int) | |||||||||
| S(hb_ext) is hydrogen bondings external | |||||||||
| S(vdw_ext) is van der waals interactions external | |||||||||
| S(hb_int) is hydrogen bond internal | |||||||||
| S (int) is internal bondings | |||||||||
| Molecule | No. of Hydrogen bond | Protein | Atoms | Bond length molecule (Å) |
Docking score (K.J/mol) |
| Iturin A | 4 | GLU215 (OE2) | 14(H) | 1.677 | -16.52 |
| - | - | LEU216 (O) | 15(H) | 1.634 | - |
| - | - | LYS217 (HZ3) | 10 (O) | 2.561 | - |
| - | - | GLU237 (OE2) | 18 (H) | 2.427 | - |
HZ represents hydrogen zeta
CONCLUSION
Ongoing work in the field of insecticide production has been done on S.litura; the question of raising the level of insect tolerance to the usable insecticides must, therefore, be solved. The protease in the cytoplasm to vacuole transport (Cvt) and autophagy pathway was selected based on this background. It reflected a measured goal for the drug target. Molecular simulation is typically the tool of choice, in the absence of an experimentally defined structure. 3-D Protease structure was developed using 2D1I. The model created further enhances simulations of molecular dynamics. In the reconstruction of MD, it is possible to convey that the general disposition of the atom remained remarkably unchanged as part of the reenactment of the structure and did not undergo any significant alteration. This model was eligible to use certain approval strategies like PROCHECK, ERRAT, envision a situation in PROSA and validate in VERIFY 3D. Additionally, the optimized model was subjected to atomic dockage. Iturin A's molecular docking against the protease paradigm showed strong in vitro inhibitory activity. The accessibility of this manufactured model will clear a route for inquires for structuring clinically selected inhibitor (iturin A) against Spodoptera therapeutics for sustainable agriculture.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
All the authors declared no conflict interest.
ACKNOWLEDGEMENTS
The authors wish to express our thanks to the Suranaree University of Technology for providing support. Also, we would like to express our special thanks to the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, Ministry of Education for support fellowship to Postdoctoral full-time researcher Dr. Narendra Kumar.

