
The Development and Progress in Machine Learning for Protein Subcellular Localization Prediction
Abstract
Protein subcellular localization is a novel and promising area and is defined as searching for the specific location of proteins inside the cell, such as in the nucleus, in the cytoplasm or on the cell membrane. With the rapid development of next-generation sequencing technology, more and more new protein sequences have been continuously discovered. It is no longer sufficient to merely use traditional wet experimental methods to predict the subcellular localization of these new proteins. Therefore, it is urgent to develop high-throughput computational methods to achieve quick and precise protein subcellular localization predictions. This review summarizes the development of prediction methods for protein subcellular localization over the past decades, expounds on the application of various machine learning methods in this field, and compares the properties and performance of various well-known predictors. The narrative of this review mainly revolves around three main types of methods, namely, the sequence-based methods, the knowledge-based methods, and the fusion methods. A special focus is on the gene ontology (GO)-based methods and the PLoc series methods. Finally, this review looks forward to the future development directions of protein subcellular localization prediction.
1. INTRODUCTION
The prediction of protein subcellular localization is an important research direction in proteomics and molecular cell biology, exerting an extensive and profound influence on protein function annotation, drug target discovery, and drug design [1-3]. Using viral proteins as an example, understanding the subcellular localization of the SARS-CoV-2 viral proteins in the host cells can promote the development of antiviral drugs, which are important in preventing the COVID-19 pandemic. Using plant proteins as another example, a study [4] analyzed the subcellular localizations of 13 enzymes and regulatory proteins in stably transformed Arabidopsis thaliana, which provides fundamental insights into the relative functional contributions of each individual component and a critical first step for further bio-design. Furthermore, for microbial protein, Peabody, M. A. et al [5] used a bacterial and archaeal protein subcellular localization prediction tool to detect water quality. More importantly, understanding the subcellular localization of human proteins has profound clinical significance. For a leading-edge example, it can promote biomarker discovery, a process requiring the information of protein subcellular localization and translocation, and potentially contribute to cancer diagnosis [6]. To be specific, Xue et al [7] attempt to screen colon cancer marker proteins in clinical settings to help with early screening, diagnosis, and monitoring of metastasis and recurrence of cancer. For other diseases, Higa et al [8] reveal the role of subcellular localization of Arid5a protein for the regulation of inflammatory responses to find new targets for the treatment of immune diseases.
In this review, we have explored and integrated a large body of literature in recent years in this area and have identified two major knowledge gaps that we are currently facing. After that, we present the most representative protein subcellular localization predictors based on three main types of methods, namely, the sequence-based methods, the knowledge-based methods, and the fusion methods. Specifically, we focus on a GO-based method developed by Wan et al [9-16] and a novel fusion method, i.e., the pLoc series method, developed by Chou et al [17-35]. Moreover, we have quantitatively analyzed, compared and visualized the performance data of several representative predictors. Finally, we give our own perspectives on the future development directions of protein subcellular localization prediction.
1.1. Current Gaps
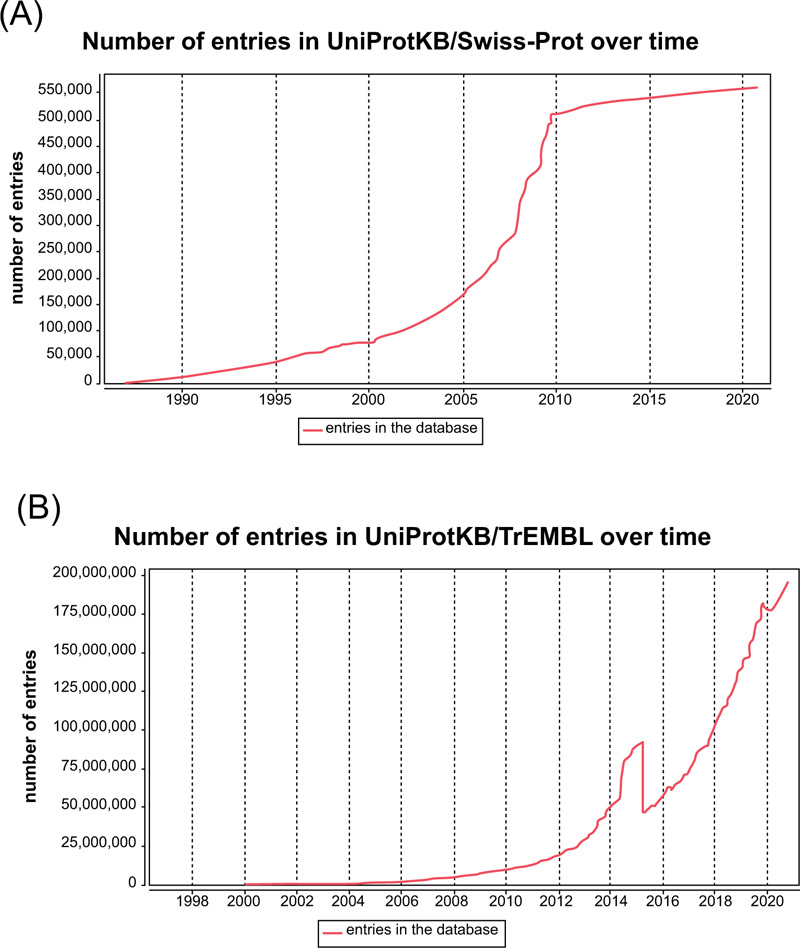
Currently, there are two prominent obstacles in protein subcellular localization prediction. Firstly, the number of novel and unreviewed proteins grows rapidly from 10,867,798 (05/08/2010) to 195,104,019 (10/07/2020) Fig. (14a), with a net increase of 184,236,221 in less than ten years [36]. In contrast, the number of reviewed proteins grows from 517,100 (05/08/2010) to 563,552 (10/07/2020) (Fig. 1b), only with a net increase of 46,452 in the same period [36]. The number of unreviewed proteins is 40 times the number of reviewed proteins. As the number of unreviewed proteins grows exponentially, the current protein prediction methods are not efficient enough to review these new proteins. Therefore, it is necessary and urgent to develop high-throughput computational methods to deal with these large-scale unreviewed proteins. Of all the potential approaches, machine learning is the most promising one.
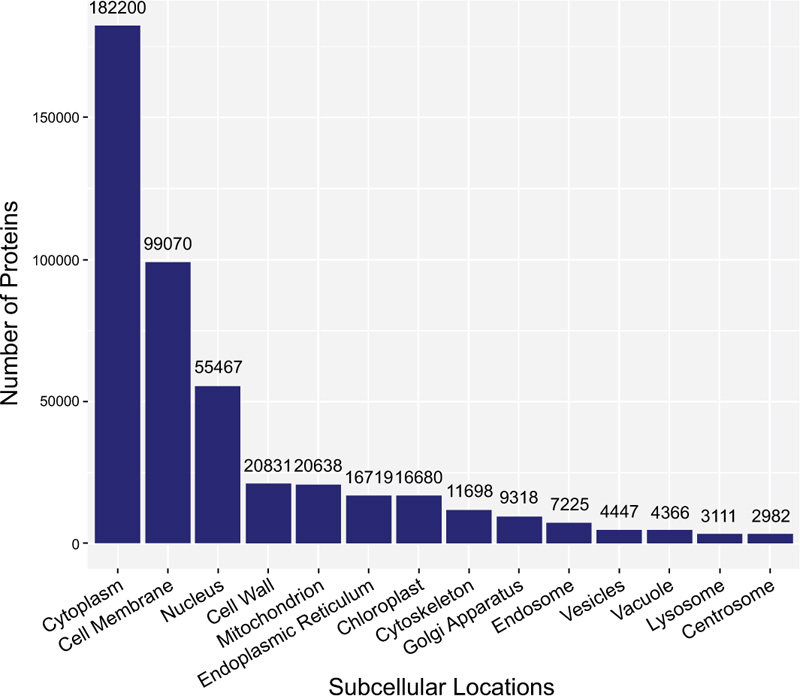
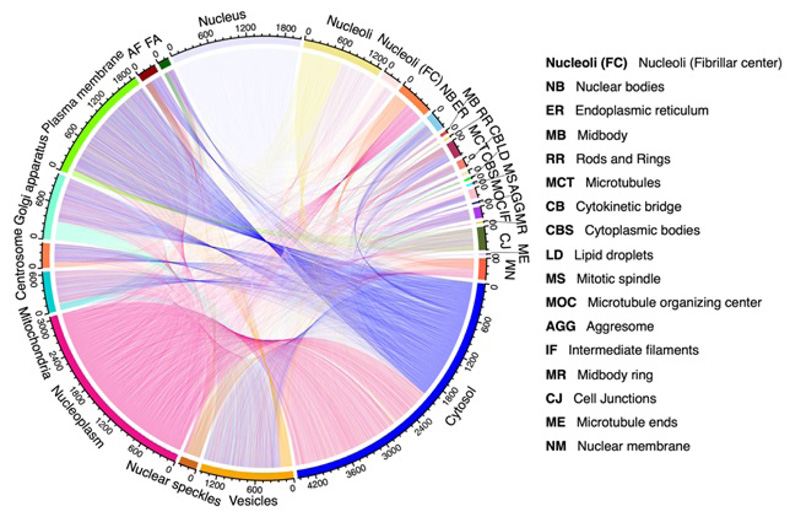
Secondly, it is known that proteins exist in some specific subcellular locations (Fig. 2), and there are already many existing predictors for single-label proteins. However, proteins do not stay at only one site; instead, they can simultaneously reside at, or move between, two or more subcellular locations [37-40] (Fig. 3). Previous studies have demonstrated that these proteins at multiple locations play an irreplaceable role in the metabolic system [41]. Therefore, many multi-label predictors have been developed to tackle this problem in the past decade.
2. SEQUENCE-BASED METHODS
Sequence-based methods make use of the amino acid sequence of the query protein to find the correlation between the sequence and subcellular localization, which can be further divided into three categories. The first category is the composition-based methods, which focus on the relationship between subcellular localization and amino acid sequence information and can be further classified into 3 types: the amino-acid composition (AA) method [43, 44], the amino-acid pair composition (PairAA) method [44], and the gapped amino-acid pair composition (GapAA) method [45, 46]. All these three types of methods are based on the frequency of amino acids. To be more specific, PairAA counts the number of occurrences of AA pairs in a protein, and GapAA counts the frequencies of AA pairs whose residues are separated by gaps. The second category is the sorting signal-based methods, which can recognize the N-terminal sorting signals in amino acid sequences, including the information where the protein will be transported, to predict protein subcellular localization. The third category is the homology-based methods. As homologous sequences are more likely to reside in the same subcellular location, the query sequence is searched against a protein database to determine whether this sequence has known homologs [47, 48]. If one or more homologs are identified, the subcellular location of the query sequence will be the same as for the homolog(s).
2.1. Pseudo-Amino-Acid Composition Features
A lot of recent studies focused on one composition-based method named the Pseudo-amino-acid composition features (PseAA) method proposed by Chou et al. [49, 50]. Based on the AA-composition features, PseAA can determine many biochemical properties of the sequence, such as hydrophobicity, hydrophilicity, and side-chain mass of amino acids from protein sequences by a sequence-order correlation factor. Compared with AA, PairAA and GapAA, PseAA combines the basic normalized AA models and replaces the co-occurrence frequencies with biochemical properties of amino acids. In addition, PseAA integrates the sequence-order information from the biochemical properties of amino acids to construct a dense feature vector in a very low dimensional space [41], which is different from PairAA and GapAA, which formulate a sparse feature vector in a high-dimensional space. Importantly, PseAA can also be easily modified to incorporate more biochemical properties, leading to the development of a number of improved PseAA models [51-60]. There are many classifiers [52-60] based on PseAA that have been proposed for protein subcellular localization.
2.2. Chou’s 5-Step Rule
For the practical design of protein subcellular localization predictors, Chou originally proposed the “5-Steps Rule” or “5-Step Rules” in 2011 [61] (hereafter referred to as the 5-steps rule). The 5-steps rule aims to enable the development of a practical and reliable statistical predictor based on genomic or proteomic data. The 5 steps are as follows [61]: 1) Construct or select a valid benchmark dataset to train and test the predictor, 2) Formulate the sequence with an effective mathematical algorithm that can truly reflect intrinsic correlation with the target to be predicted, 3) Develop a powerful algorithm (or engine) to predict the subcellular protein localization, 4) Perform cross-validation tests to evaluate the accuracy of the predictor, and 5) Establish a user-friendly web server for the predictor that is accessible to the public. Till now, the above 5-steps rule has been used by many scientists in developing various predictors for proteomic or genomic analyses. Chou’s 5-step rule has many merits [61]: 1) crystal clear in logic development, 2) completely transparent in operation, 3) reported result easy to be repeated by other investigators, 4) high performance in simulating other sequence-based methods, and 5) very convenient to be used by the majority of experimental scientists.
3. KNOWLEDGE-BASED METHODS
Knowledge-based methods are different from sequence-based methods because they extract the features of query protein from knowledge-related databases, such as the Gene Ontology (GO) database [41], the Swiss-Prot keywords database [62, 63], or the PubMed abstracts database [64]. These methods use annotations of a protein to correlate the query protein with the subcellular localization. Among all knowledge-based methods, the GO-based method is the most popular one, which makes use of well-organized biological knowledge about genes and gene products.
In more detail, the GO database is a set of standardized vocabularies that annotate the function of genes and gene products across various species. In the GO database, the annotations of gene products are organized in three related ontologies: cellular components, biological processes, and molecular functions.
3.1. The Legitimacy of Using Gene Ontology Information
Four solid theoretical foundations are proposed to support the legitimacy of gene ontology information utility. Firstly, the GO-based method is not a simple table-lookup that converts the annotation into another format and GO terms could not be used to determine the subcellular locations of proteins directly, as some proteins do not have annotations regarding cellular components while others could have multiple annotations regarding cellular components in the GO terms. Thus, some proteins do not have information regarding subcellular localization, while others might have multiple subcellular localizations. According to Chou et al [65], proteins with annotated subcellular localization information in the Swiss-Prot database may still be marked as “cellular component unknown” in the GO database. Secondly, Mei et al [66] did extensive tests on the Multiloc [67], BaCelLo [68], and euk-mPLoc [49] datasets and showed that not only cellular components but also molecular functions and biological processes in the GO terms play significant roles in estimating the kernel weights of the proposed classifier and contributing to the final prediction accuracy of the model, making GO-based methods outperform sequence-based methods. Thirdly, GO-based methods remarkably outperform Briesemeister’s methods [69]; the latter use only homologous transfer and a basic local alignment search tool (BLAST) which might not be sufficient for reliable prediction. Fourthly, the legitimacy of using GO information is also supported by Chou et al [70], who suggested that as long as the input of query proteins contains only sequence information without any GO annotations, the output is the subcellular localization information; this method should be regarded as equally legitimate for subcellular localization.
3.2. Single-label Predictors
Proteins that reside in only one subcellular location represent most cellular proteins. Therefore, predicting the subcellular localization of single-label proteins is important. A GO-based predictor, GOASVM, is one of the most representative GO-based predictors that have laid a critical foundation for further improvement and development of various other protein subcellular localization predictors.
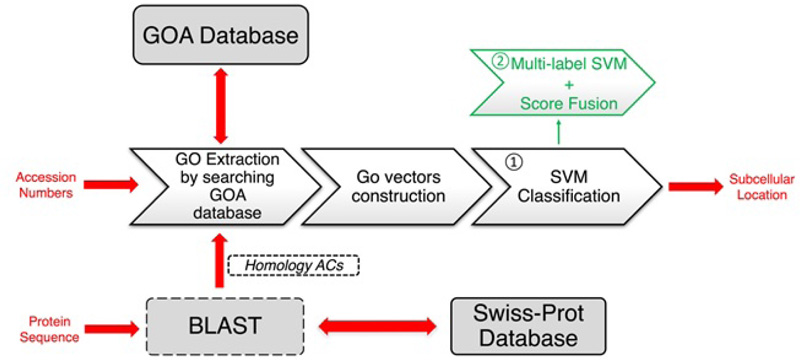
The GOASVM predictor takes only the GO information as the input features and adopts a successive search strategy to make sure it can be applied to novel proteins. We illustrated the strategy by generating a workflow of GOASVM. As shown in Fig. (4), GOASVM uses either accession numbers (ACs), which is a unique identifier given to a protein sequence, or amino acid (AA) sequences as input and extracts the GO information from the GO database. The prediction process is divided into two stages: feature extraction (vectorization) and pattern classification. For the former, the query proteins are “vectorized” to high-dimensional GO vectors, and the elements of these vectors are determined [41]. For the latter, the GO vectors are classified by one-vs-rest linear support vector machines (SVMs). If ACs are available, the GO training vectors will be created based on ACs. However, if only the AA sequences are known, then ACs of homologs can be used for training the SVM, and the GO training vectors will be created by using ACs of homologs only. The performance of GOASVM can be evaluated by the leave-one-out cross-validation (LOOCV).
Compared with other state-of-the-art GO-based single-label predictors, such as ProLoc-GO and Hum-Ploc [41], GOASVM performs best regardless of whether input data is ACs or AA sequences. For instance, GOASVM achieves accuracies of 94.68% and 94.61% in the EU16 dataset when AA sequences or ACs are used as inputs, respectively, whereas other predictors achieve only accuracies of 89.0% and 85.7% when AA sequences or ACs are used inputs, respectively.
3.3. Multi-Label Predictors
As mentioned above, single-label protein subcellular localization prediction is far from enough to completely predict the subcellular localization of all proteins. Therefore, the development and evaluation of multi-label predictors for proteins with multi-locations are essential. Here, we present seven efficient multilabel classifiers that are commonly used for protein subcellular localization prediction, i.e., mGOASVM [10], AD-SVM [11], mPLR-Loc [12], SS-Loc [16], HybridGO-Loc [13], RP-SVM [14], and R3P-Loc [15]. All these seven predictors use GO information as input features for protein subcellular localization prediction.
3.3.1. Basic Multi-Label Predictors
As an improved version of the original GOASVM, mGOASVM was developed for multi-label protein subcellular localization prediction with three major improvements (Fig. 4). For feature extraction, mGOASVM adopts more than one homologous protein from the GO database, enabling the retrieval of relevant GO terms to form a more informative GO subspace. Moreover, mGOASVM adopts a new multi-label SVM classifier which can effectively deal with datasets containing both single-label and multi-label proteins.

As the simple binary relevance method is used to deal with multi-label problems in mGOASVM, a large number of false positives will exist in its predictions. To tackle this problem, an adaptive decision multi-label predictor named AD-SVM, which uses an adaptive decision scheme based on the maximum score of one-vs-rest SVM, was developed. It effectively reduces the ratio of false positives while exerting little influence on the prediction accuracy.
Another predictor named mPLR-Loc was also developed using the same adaptive decision scheme as AD-SVM, the logistic regression (LR) classifier. One important feature of mPLR-Loc is that the LR posterior scores used in the model are probabilistic, which may provide better biological meaning when compared with the AD-SVM method.
3.3.2. Mining Deeper on GO for Protein Subcellular Localization
One important improvement of basic multi-label predictors is the extraction of deeper GO information. Two predictors, namely SS-Loc and HybridGO-Loc, extract semantic similarity (SS), a deeper GO information reflecting the relationships among the GO terms, for protein subcellular localization prediction.
In detail, SS-Loc extracts GO information and SS from GO terms to construct the similarity vectors, which are inputs for the multi-label SVM classifier. Based on this, HybridGO-Loc extracts the features of GO term frequency and GO SS to generate hybrid vectors. As shown in Fig. (5), the input for the HybridGO-Loc is GO hybridized vectors. The outstanding performance (in 3.3.4) of HybridGo-Loc further proves the hybridization of GO frequency and GO SS is a reasonable approach.
3.3.3. Ensemble Random Projection for Large-Scale Protein Subcellular Localization
Another improvement of basic multi-label predictors is the dimensionality reduction of GO vectors. Generally, thousands of GO terms formulate high-dimensional GO vectors during the GO vector construction, which may contain redundant or irrelevant information, causing the overfitting of predictors. To tackle this problem, Wang et al [14, 15] developed two dimensionality reduction methods, namely, RP-SVM and R3P-Loc, which can apply random projection (RP) to construct an ensemble of multi-label classifiers. Ensemble RP can project GO vectors onto lower-dimensional space and thus form a lower-dimensional GO vector, which can subsequently be classified by an ensemble of one-vs-rest multi-label classifiers (Fig. 6). The difference between RP-SVM and R3P-Loc is that the former utilizes a multi-label SVM classifier and the ProSeq database, while the latter uses a multi-label ridge regression classifier and a ProSeq-GO database.
ProSeq and ProSeq-GO are two compact databases. The former is a sequence database created from the Swiss-Prot database, and the latter is a GO-term database created from the GO database. These compact databases could reduce the memory consumption by 39 times while at the same time keeping the performance almost unaffected.
3.3.4. Properties and Performance of Multi-label Predictors
We integrated and summarized the properties of multi-label predictors, as shown in Fig. (7). Since the existence of null GO vectors will reduce the performance of predictors, all the predictors discussed in this review adopt a new successive search strategy that can avoid the null GO vectors and thus avoid their negative impact. All the predictors except SS-Loc use term frequencies for constructing feature vectors, which could improve the performance of these predictors. Regarding classifier improvement, AD-SVM and HybridGO-loc use an adaptive decision scheme based on the multi-label SVM classifier used in mGOASVM, while mPLR-Loc and R3P-Loc use multi-label penalized logistic regression and ridge regression classifiers, respectively [41]. To mine deeper GO information, HybridGO-Loc and SS-Loc employ the GO SS for classification. In terms of dimensionality reduction, RP-SVM and R3P-Loc adopt random projection ensemble, which can reduce the high dimensionality of GO vectors and boost the prediction performance.


Based on mGOASVM, the other developed predictors have made different improvements in aspects, such as deeper feature extraction, dimensionality reduction, and refinement of multi-label classifiers. We calculated the performance parameters, and the results are represented in Fig. (8). The performance of these predictors is evaluated by seven metrics, i.e., accuracy, precision, recall, F1, HL, OLA, and OAA. It is clear that except for recall and OLA, HybridGO-Loc performs the best among all the predictors, demonstrating that two metrics, GO and SS, are complementary to each other. Regarding OLA and recall, SS-Loc has the best performance, which further proves that mining deeper GO information (i.e., semantic similarity information) is critical for improving the predictor performance.
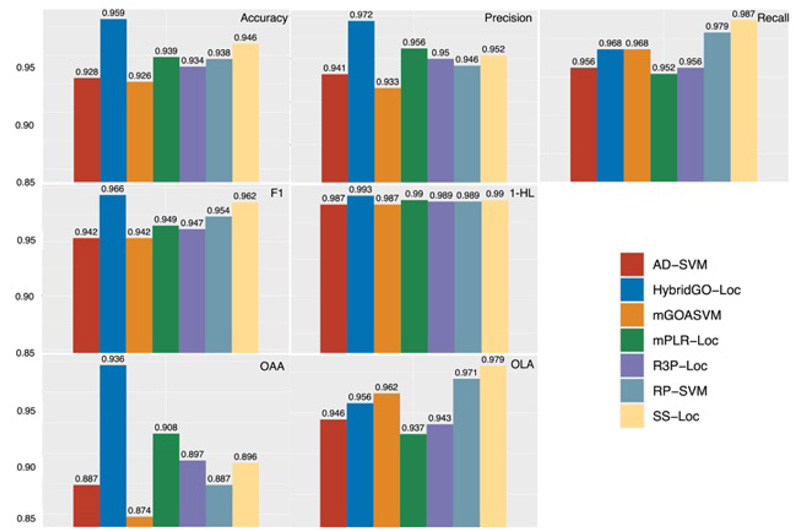
3.3.5. Interpretability of Prediction Result
Another urgent question remaining in protein subcellular localization prediction is that the prediction results are usually hard to be interpreted. However, these predictors should not only provide the subcellular localization information of new proteins but also clarify the underlying mechanisms why these proteins are located in specific sites. Wan et al [71-74] have developed two classifiers to tackle this problem, which they named mEN and mLASSO.
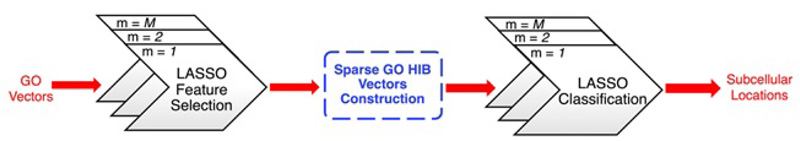
Absolute shrinkage and selection operator (LASSO) (Tibshirani, 1996) is an L1-regularized linear regression model. As shown in Fig. (9), Wan et al have made a LASSO feature selection from the ProSeq-GO database (a compact database as mentioned in 3.3.3), which can be used for the training of normal GO vectors with irrelevant features (or GO terms) removed. They used some of the depth-dependent GO hierarchical information of essential GO terms, representing the relationship between different GO terms, to get a spare GO hierarchical information-based (HIB) vector. In the final step, they applied the LASSO classification to the HIB vectors and got output regarding protein subcellular localizations.
One crucial problem for LASSO is that the results from LASSO tend to give very sparse solutions, causing the missing of some important information from the feature list. Therefore, a multi-label elastic net (EN)-based classifier is designed to overcome this shortcoming. One convex combination can yield sparse representations in EN, which is similar to those in LASSO, and reveal correlated features that can be selected or deselected together [72]. Actually, LASSO can be regarded as a special case of EN. In short, compared to LASSO, EN will select correlated features together, thus causing more essential GO terms to be selected.
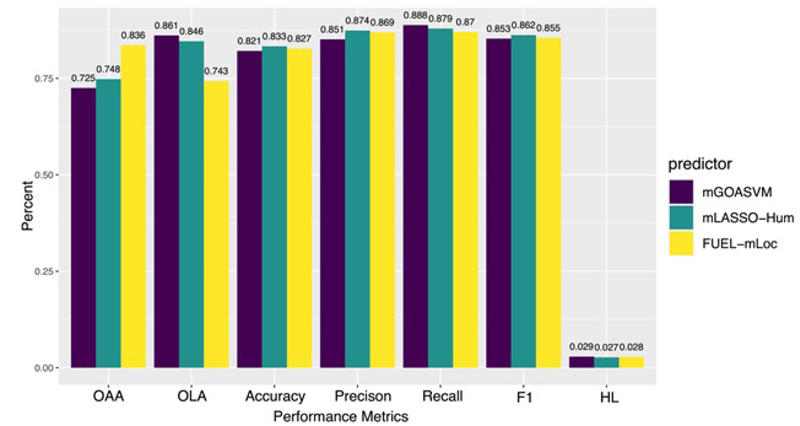
By using mEN and mLASSO, Wan et al developed four multi-label predictors, which are mLASSO-Hum [71], Gram-LocEN [72], Mem-Men [73], and FUEL-mLoc [74]. The overall performance of these predictors is comparable to that of mGOASVM (Fig. 10). However, the key contribution of mEN and mLASSO endow the predicting result with biological significance. Using mLASSO-Hum, 87 out of more than 8000 GO terms are found to play significant roles in determining the subcellular localization. Using one-vs-rest EN classifiers, 162 and 245 out of more than 8,000 GO terms are selected for Gram-positive and Gram-negative bacteria, respectively. These results are all consistent with biological annotations, indicating that the key GO terms have higher weights in determining the corresponding protein subcellular location. In addition, they also proved that the GO terms derived from cell components, molecular functions, and biological processes could contribute to protein subcellular localization prediction, implying these predictors can provide interpretations for the prediction results.
4. FUSION METHODS
For the fusion methods, Chou et al combined GO information and PseAA into a new predictor series. Recently, their research about protein subcellular localization prediction can be generally classified into three series: 1) pLoc-mX [17-23], 2) pLoc_bal-mX [23-28], and 3) pLoc_deep-Mx [29-35], where X denotes “Euk” (eukaryotic), “Hum” (human), “Animal”, “Plant”, “Virus”, “Gneg” (Gram-negative bacterial), “Gpos” (Gram-positive bacterial) proteins, respectively. “pLoc” denotes “predicted subcellular localization”, “m” denotes “multi-label”, “bal” denotes “balancing” and “deep” denotes “deep learning”.
4.1. Methods
Predictors, including pLoc-mHum, pLoc_bal-mHum, and pLoc_deep-mHum, for the human protein database, are evaluated. The other datasets share very similar research methods. As the original prototype, pLoc-mHum incorporates the optimal GO information into Chou et al’s general PseAAC and inputs it into the ML0GKR classifier to predict the subcellular protein localizations. The function of the optimal GO information reduces the general PseAAC vector’s dimension.
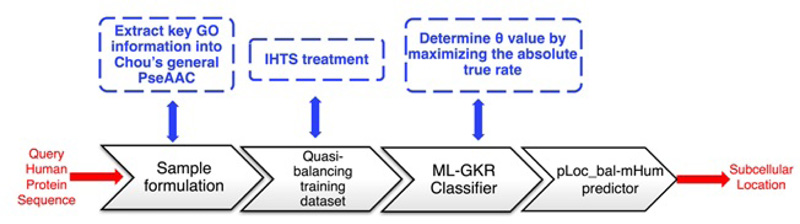
As a predictor improved from pLoc-mHum, pLoc_bal-mHum tackles the biased consequence of pLoc-mHum, which is caused by an extremely skewed benchmark dataset, i.e., the number of total proteins in different organelles is very different. For example, according to the benchmark dataset used in the study by Xiao et al [22], pLoc-mHum predicted synapse protein number is 22, endosomal protein number is 24. In contrast, nuclear protein number is 1021, and cytoplasmic protein number is 817. To solve this problem, pLoc_bal-mHum was applied to IHTS (Inserting Hypothetical Training Samples) to create a Quasi-balancing training dataset that adds some reasonable samples into the smaller subsets to make the skewed benchmark dataset more balanced. We extracted the main elements of pLoc_bal-mHum and generated a workflow, as shown in Fig. (11).
By combining the deep-learning techniques, Chou et al developed pLoc_deep-mHum, which is a CNN-BiLSTM neural network model that includes one convolution layer and one BiLSTM block. The superiority of this model is that the CNN convolution layer can extract the maximum amount of information from human protein features, which will be used as input for BiLSTM. BiLSTM, as a classifier, can filter the information through the CNN layer. Finally, the vectors from CNN are transformed into probability to define the class of each output.
4.2. Performance and Comparison
To quantitatively evaluate these three series of multi-label predictors, Chou et al used two sets of metrics: one for its global accuracy and the other for its local accuracy. Global accuracy is defined by a set of five metrics [69]: aiming, coverage, accuracy, absolute true, and absolute false (Fig. 12). For the absolute_false metric, the smaller the value, the better the performance; For all other metrics, the higher the value, the better the performance. Chou et al’s four intuitive metrics, namely, sensitivity, specificity, accuracy, and Mathew’s correlation coefficient, are used to evaluate the local accuracy of these three multi-label predictors [75, 76] (Fig. 13).
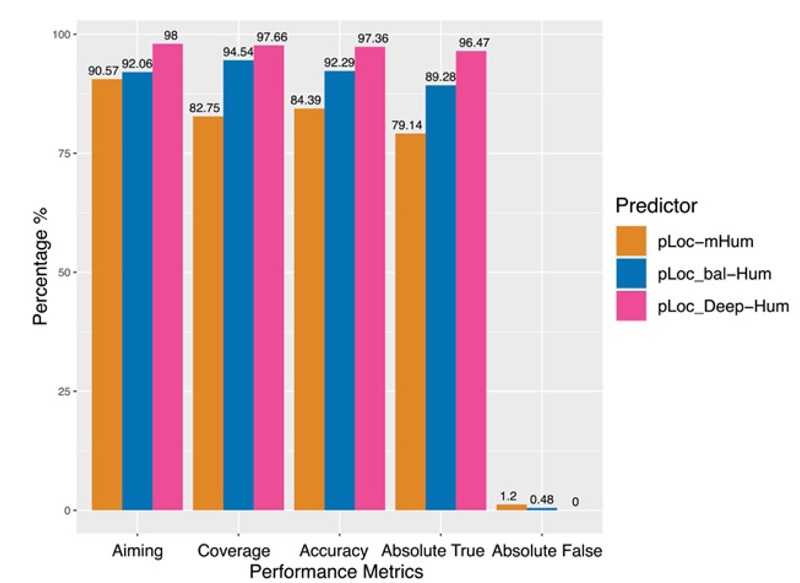
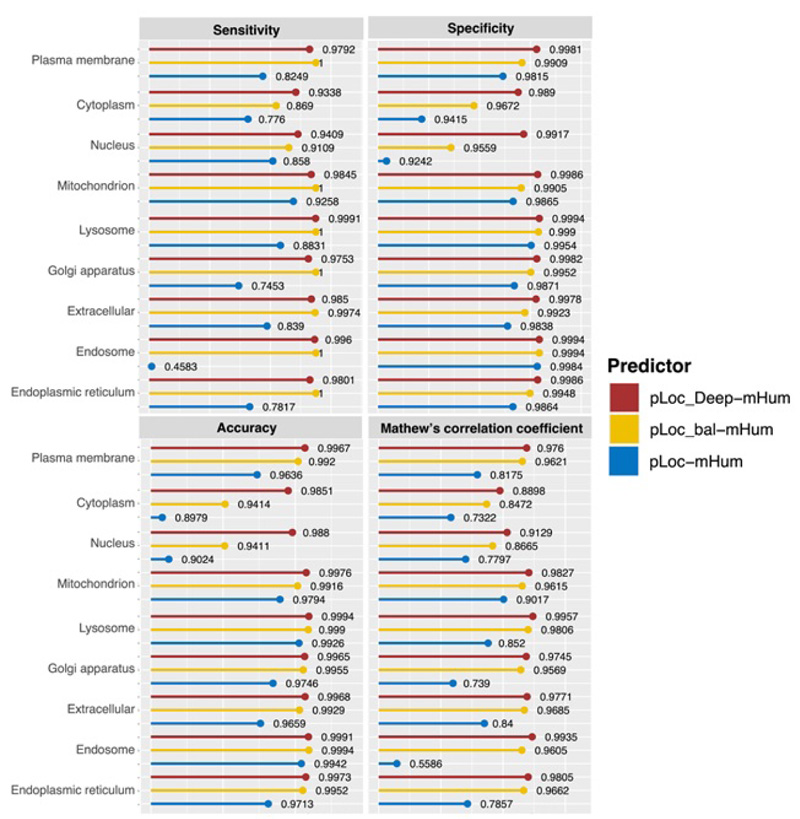
It is clear that the pLoc_Deep-Hum performs the best regarding the five metrics in global accuracy and a large portion of specific organelles, which reflects the superiority of the pLoc-Deep-X series. For pLoc_balHum, it performs much better than pLoc-mHum. However, except for some metrics in specific organelles, such as the sensitivity in the cytoplasm and the accuracy in the endosome, it falls behind the pLoc_Deep-Hum among global accuracy and most local accuracy. For pLoc-Hum, it is more like the base of the other two predictors.
Moreover, an interesting finding is that the improvement of the algorithm from pLoc-Hum to pLoc_bal-Hum or pLoc_Deep-Hum can significantly improve the prediction performance in certain subcellular locations, such as nucleus, endosome, and cytoplasm. However, other locations, such as mitochondrion, have shown better results from pLoc-Hum, so the improvement of the algorithm has less impact on their prediction performance.
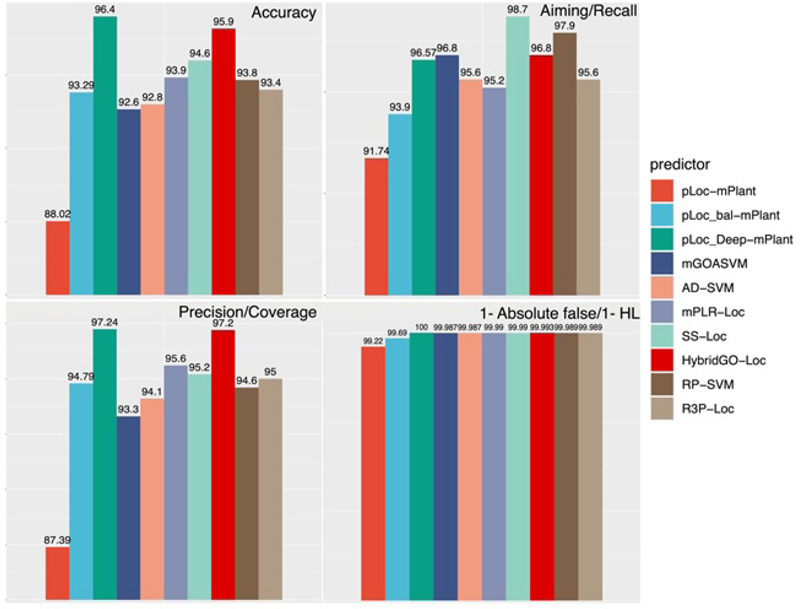
Lastly, we also calculated and compared the performance of all these predictors based on the PLoc series with GO-based methods (Fig. 14). For the rationality of comparison, we selected a plant database that has been tested by all these predictors and selected four common metrics, which are accuracy, recall/aiming, precision/coverage and HL. From the results, we concluded that among all predictors, pLoc-deep-mPlant performs best in terms of accuracy, precision, and absolute false rate. These results prove the superiority of predictors based on deep learning techniques. SS-Loc performs best regarding the recall, and semantic similarity has a significant effect on this metric. As suggested from our performance analyses, for future directions, one path would be to fully combine the advantages of the two kinds of methods (i.e., GO-based methods and PLoc series). Predictors based on deep learning techniques and combined GO features and SS features like HybridGO-Loc can be designed to further improve the overall prediction performance.
CONCLUSION
In this review, we introduce the development and progress of machine learning in protein subcellular localization prediction. We not only include an explanation for detailed steps of the predictors but also compare the performance differences between different predictors, which can provide a quick and powerful reference for scholars interested in protein subcellular localization prediction and are looking forward to seeking a breakthrough. Especially, we focus on the multi-label predictors of GO-based methods and PLoc-mX, PLoc-bal-mX, Ploc-deep-mX methods, which are all state-of-the-art predictors belonging to the fusion method. The mGOASVM method is the base of GO-based methods, many of which are improved in certain steps on the mGOASVM. We can summarize that AD-SVM and mPLR-Loc improve the classifier, SS-Loc and HybridGO-Loc improve the deep feature extraction, RP-SVM and R3P-Loc improve the dimensionality reduction, and mLASSO and mEN provide the interpretability of prediction results. For the ploc series, the foundation is PLoc-mX; PLoc-bal-mX equips a balancing training dataset, and ploc-deep-mX takes advantage of deep learning techniques. In practical applications, we should choose appropriate predictors according to different research purposes to achieve the optimal prediction effect. For example, using the localization of cancer marker proteins for early diagnosis of cancer requires high sensitivity, so the improved classifier property is particularly important. While for the inflammatory response regulation, different regulatory factors are induced at different subcellular locations to mediate different pathways, resulting in completely opposite regulatory effects. Therefore, we need more refined location features. In this case, the deep feature extraction property should be the priority. In a longer-term clinical significance, accurate prediction of protein subcellular localization can contribute to the design of new drugs, which have the potential for curing diseases and benefiting all humankind.
LIST OF ABBREVIATIONS
| AA | = Amino-acid |
| GapAA | = Gapped Amino-acid |
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

