
A Study of Bio-inspired Computing in Bioinformatics: A State-of-the-art Literature Survey
Authors Info & Affiliations
Abstract
Background:
Bioinspired computing algorithms are population-based probabilistic search optimization approaches inspired by biological evolution and activity. These are highly efficient and can solve several problems based on human, chimpanzee, bird, and insect behavior. These approaches have been proposed by the scientific community over the last two decades for common application to solving bioinformatics design problems.
Materials and Methodology:
The advanced search boxes in databases such as PubMed, WoS, Science Direct, IEEE Xplore, and Scopus to conduct this research. Keywords such as "machine learning," "bioinspired computing," "DNA sequence optimization," and "bioinformatics" were used with OR and AND operators. Journal and conference articles were the two types of articles focused on, and other reports and book chapters were removed using the search engine's parameters.
Results:
Bioinspired techniques are becoming increasingly popular in computer science, electrical engineering, applied mathematics, aeronautical engineering, and bioinformatics. Parametric comparisons suggest that most classic benchmark approaches can be successfully used by employing bioinspired techniques. 56 % of studies are modification based, 30 % hybrid based, and 14 % multiobjective based.
Conclusion:
These algorithms can be used to optimize data sets in bioinformatics due to their capacity to solve real-world challenges and their ability to accurately express sequence quality and evaluate DNA sequence optimization.
1. INTRODUCTION
Transferring concepts from biology into the field of computer science led to the emergence of bioinspired computing, it evolved independently for many years. It is extended by approaches from optimization techniques, complexity theory, and probability theory. Here, we review some recent theoretical developments in bioinspired techniques and their potential applications to computer science. In this paper, a comprehensive analysis of bioinspired multiobjective optimization and its applications in bioinformatics is reviewed. To enhance the performance of modern computers and solve a broad array of optimization challenges, a variety of biological concepts and models are being incorporated into computer science. In today's reality, a highly advanced computer must use biology as a platform for evaluating the intricacies of algorithms and improving the outcomes to represent the computer's adaptability. There are several approaches to solve optimization problems, all of which necessitate significant computing resources and tend to diminish as the problem size grows. For this reason, deterministic methods are being replaced with stochastic optimization algorithms that draw inspiration from biological systems [1]. Bioinspired computation [2] is now widely used in a variety of sectors with real-world applications, including business, management, science, and engineering. Furthermore, the evolution of traditional neural networks and the introduction of deep learning technologies have influenced the design of nature-inspired algorithms to address a wide range of scientific and engineering challenges.
This study focuses on bioinspired approaches that can be applied to resolve issues in the real world for human problems. Various instances of bringing bioinspired approaches to bioinformatics and related applications are now available in the literature. Biologists and healthcare professionals may benefit from DNA sequencing technology for a variety of tasks, such as molecular cloning, breeding, locating pathogenic genes, and comparative and evolutionary research. The genetic information of life is carried by DNA; it promotes the expansion of biological development and the function of living systems. Currently, bioinspired machine learning algorithms are extensively used in sequence data analysis and have a wide variety of application possibilities in terms of improving data processing capabilities and providing useful biological information. The bioinspired machine learning techniques based on DNA sequences in bioinformatics have introduced the sequencing technology development process, DNA sequence data structure, and various sequence encoding approaches [3-6] in bioinspired-based machine learning algorithms. In addition, we have reviewed these models, which are an automated estimation of the disease, which is diagnosed using machine learning and bioinspired computing techniques. Improving screening by allowing rapid diagnosis on a large scale when physical testing cannot achieve it with techniques like machine learning, which can use the ample amounts of data produced by many laboratory tests, clinical symptoms, and radiological scans to improve diagnosis will be extremely effective [7]. To anticipate infected individuals, machine learning techniques can use existing quantitative and qualitative data [8, 9]. The results of this approach can help doctors make better decisions during diagnosis and treatment, leading to an improvement in the fight against various diseases. Finally, we examine some of the fundamental bioinspired computing algorithms applied in bioinformatics and discuss how they are applied to improve machine learning approaches with their advantages and disadvantages.
The following is how the remaining parts are organized: The classification of bioinspired computing methodologies is presented in Section 1.1, followed by overviews of bioinformatics and various problem types in Section 1.2, an article search process in Section 2, and a review of the literature on bioinspired computing for various bioinformatics issues in Section 3. Section 4 highlights bioinformatics' unresolved research problems and potential future research areas. Section 5 provides an overall summary of the literature survey with discussion, and Section 6 concludes with a conclusion and an updated list of really useful references.
1.1. Bioinspired Computing
Recently, several bioinspired computing optimization strategies have been proposed by researchers. These algorithms are distinct from other metaheuristic algorithms in that they are based on biological behavior, especially that of plants, animals, and insects. In today's interdisciplinary field of theoretical study, bioinspired computing is gaining interest because most bioinspired algorithms use probabilistic and stochastic search approaches to solve large-scale optimization problems with near-optimal solutions. As traditional mathematical optimization techniques frequently fail due to solutions becoming stuck in local optima, derivative-free metaheuristic global optimization strategies have been developed. The primary purpose of this research is to introduce the basic bioinspired methodologies identified in the literature and their applications in bioinformatics. This study examined a few of the algorithms listed below that were inspired by biological systems. Genetic Algorithm (GA), Particle Swarm Optimization (PSO), Ant Colony Optimization (ACO), Bat Search (BS) algorithm, Evolutionary Algorithm (EA), Cuckoo Search (CS), Grey Wolf Optimization (GWO), Whale Optimization Algorithm (WOA), Slime Mold Algorithm (SMA). The reason for the increased interest in the field is that it overcomes the limitations of standard models. This method is often employed in both educational and professional settings. It is believed that as human social requirements get more complicated, a slew of new bioinspired strategies will arise.
1.2. Bioinformatics
Bioinformatics is a key discipline that combines computer science, mathematics, statistics, engineering, and biology to help answer biological issues. To designate the study of informatics processes in biotic systems, Hogeweg and Hesper introduced the word “bioinformatics” in 1970 [10]. Modeling biological systems and their use in bioinformatics are the main objectives of bioinformatics, sometimes referred to as “computational biology”. Its primary goals are to develop new algorithms and evaluate connections between biological data in a wide range of applications, including figuring out how genes and proteins work, establishing evolutionary relationships, and predicting the three-dimensional structure of proteins using computer software and particular algorithms. Using DNA and amino acid sequence analysis, the area of bioinformatics known as “gene-based taxonomy and phylogeny” (also known as “genosystematics”) establishes genetic links between various species. The analysis of mutation spectrum complexity and its patterns in DNA or protein sequences [11], the study of genome structure, the identification of damaged genes that are expressed, functional genomics, and genome evolution research are a few examples of the applications of bioinformatics techniques. The two main databases used to store a significant portion of this data were BLAST (Basic Local Alignment Search Tool) and FASTA (stands for fast-all or FASTA) [12], the latter of which went on to become among the most effective tools in molecular biology research. The current databases for evaluating gene and protein structures are examined. The researchers produced their findings by studying biological data using computational technologies. Finally, we may define bioinformatics as the linguistics section of genes. Like linguists, those working in bioinformatics search for patterns in DNA, RNA, and protein sequences (s).
1.2.1. Bioinspired ML in Bioinformatics and Applications
The exponential growth in the size and rate of capture of biomedical data during the “big data” age is posing a challenge to traditional analysis methods. Deep learning, a subset of machine learning methods with biological origins, promises to use massive data sets to find hidden patterns and make precise predictions. Machine learning has a lot of potential for the analysis of biological data sets, as is known. Building complex models that reveal their underlying structure theoretically enables greater exploitation of the accessibility of increasingly large and high-dimensional data sets. The learned models include advanced properties, enhanced interpretability, and a better knowledge of the structure of biological data [13]. In recent days, researchers have attempted to apply bioinspired machine learning to a variety of bioinformatics domains, including DNA sequence optimization, sequence alignment, feature selection, training, neural networks, genome mining, omics, biomedical imaging, electronic health records, optimizing support vector machines, clustering application, and many others. The Basic aims and application areas of Bioinformatics are depicted below in Table 1.
| Aims of Bioinformatics | Applications Areas of Bioinformatics |
|---|---|
| 1. Organize biological data in an easy-to-use format that allows biologists and researchers to save and access current data. 2. Create software tools to aid in data analysis and management. 3. To analyze and interpret the results in a biologically meaningful manner using these biological data. 4. To aid pharmaceutical industry researchers in better understanding protein structure those contribute to the development of medicines. 5. In order to enable and assist physicians in understanding the gene architecture that will aid in the recognition and diagnosis of diseases like cancer. |
1. Locating genes by using DNA sequence data 2. Predicting the structure of RNA sequences using data. 3. Predicting the location of proteins inside the cell using protein sequence data. 4. Examining images of gene expression. 5. Gaining a better understanding of hereditary illnesses such as cancer, cystic fibrosis, and sickle cell disease. 6. Gene therapy applications in general. 7. Designing medications for better treatment and avoiding side effects, as well as in the development of a better drug delivery system. |
1.2.1.1. DNA Sequence Optimization
DNA sequence optimization is an innovative and promising technique for dealing with a variety of difficult computational problems. It is also known as molecular computing using DNA because it mixes biology, chemistry, information technology, and mathematics. So it leads to a whole new standard for computation. The purpose of the DNA sequence optimization challenge is to create a collection of unique DNA sequences that cannot be hybridized with other sequences in the set. In 1994, Adleman published the first study on DNA computing [14]. He solved a directed Hamiltonian path problem (HPP) using DNA sequence optimization techniques [15].
1.2.1.2. Sequence Alignment
In bioinformatics, “sequence alignment” is a technique for comparing different DNA, RNA, or protein sequences to identify the structural, functional, or evolutionary relationships between them. Pair-wise sequence alignment, multiple sequence alignment, and structural sequence alignment are the three forms of sequence alignment.
1.2.1.2.1. Pairwise Sequence Alignment
It is a technique for identifying regions of similarity between two sequences at a time that could have functional, structural, or evolutionary relationships.
1.2.2. DNA calculation
The length of a sequence of individual genes, larger genetic areas (i.e ., clusters of genes or operons), full chromosomes, or entire genomes can be determined by calculating the precise order of nucleotides within a DNA molecule. Calculation of the length of individual nucleotides in DNA or RNA (usually represented as A, C, G, T, and U) isolated from cells of animals, plants, or almost any other source of genetic information is performed. Quick DNA computation techniques have significantly increased biological and medical research and discovery. Choose an encoding method [16] that translates the original alphabet into strings of A, C, G, and T and then synthesizes the resulting information-encoding strings as DNA single strands to encode information with DNA. For storing biological information, DNA is an excellent medium.
1.2.3. Feature Selection
In data mining and machine learning, feature selection is a crucial procedure. The purposes of feature selection are to reduce the number of features, select those that are the most representative, and remove redundant, noisy, and unimportant features. To assess the potential feature subsets, Emary et al. [17] created a wrapper technique that combines a binary version of GWO with the fitness function k-nearest neighbor (k-NN). They compared their method with GA and PSO across three benchmark datasets. They showed improved class accuracy and faster convergence speed using their GWO-based feature selection technique.
1.2.4. Training Neural Networks
Information processing models called “artificial neural networks” (ANNs) were developed as a result of studying biological nervous systems. ANNs are often used in research and applications because of how well they can capture nonlinearity and dynamicity. However, the structure and connection weights of ANNs have a significant impact on their performance. Every new metaheuristic technique has a lengthy history of being evaluated for success by rapidly optimizing neural network connection weights. One of the most popular varieties of neural networks, MLPs, was trained by Mirjalili et al. [18] using GWO. In his research, a single hidden multilayer perceptron (MLP) network's weights and biases were maximized using GWO. The proposed training approach was contrasted with some well-known evolutionary trainers, including PSO, GA, ACO, ES, and GWO. GWO's advantage in training MLP networks was shown by comparative results based on five classifying datasets and three function approximation datasets.
1.2.5. Support Vector Machines (SVM)
SVM is a classifier and regression model that is very efficient. V. Vapnik created SVM, which has a strong mathematical foundation [19]. To enhance SVM efficiency, both the kernel parameters and the error penalty parameter C must be modified. The standard approach to solving the issue is to employ a basic or thorough grid search. However, this approach is ineffective because it takes too long to consider every possible combination. To maximize these hyperparameters, numerous researchers have looked at the application of metaheuristic algorithms [19, 20]. Recently, GWO has been used to optimize the SVM's hyperparameters. Gamma and sigma parameters in SVM were modified by Eswaramoorthy et al. [21] for categorizing intracranial electroencephalogram signals. In comparison to another classifier, the results revealed greater accuracy rates.
1.2.6. Clustering Applications
Clustering is a standard machine learning and data mining technique that divides data instances into those that share specific features [22]. For clustering problems, metaheuristic algorithms are widely used and implemented. One of the best-known metaheuristic clustering algorithms is the k-means method, and most of the metaheuristic clustering algorithms presented in the literature are alternatives to this approach. The starting centroids used for the K-means approach heavily influence its performance, and a local minimum is a highly likely trap for it. To overcome the limitations of the k-means method, based on GWO, Kumar et al. [23] designed a clustering algorithm. The fundamental idea is that each member of GWO corresponds to a set number of centroids in a dataset. They calculated the fitness by adding the squared Euclidean distance between each data point and the cluster centroids. Their tests on eight datasets showed that GWO performed better than both their metaheuristic techniques and the k-means algorithm and the results revealed greater accuracy rates [21].
1.2.7. Genome Mining
Genome mining is the technique of examining genetic information to discover naturally occurring proteins, metabolic pathways, and potential interactions [24]. It requires tools such as Computational biology and bioinformatics. A large amount of data, represented by DNA sequences, is required for the mining process and is available in genomic databases. These models are applicable in several areas of medicinal chemistry, such as the identification of natural compounds [25], where new information may be generated using data mining techniques [26, 27].
1.2.8. Omics
The growth of omics applications has been driven by technological advancements. Biological systems with complex interactions are frequently measured and studied using these methods. Numerous fields of study are covered by the term “omics,” including genomes, transcriptomics, proteomics, interactomics, metabolomics, phenomics, and pharmaco- genomics, to mention a few. Each of these areas may also have numerous subdomains, each of which calls for more skill in analytical and computational methods [28].
1.2.9. Biomedical Imaging
Machine learning has had a major influence on many naturally inspired approaches in recent years. Significant advancements in voice and image recognition have resulted from it, and it can train computer programs to outperform humans while generating artistically pleasing new pictures and images. Numerous of these tasks were thought to be insurmountable for computers to do [29] and this technology is essential for biomedical imaging.
1.2.10. Electronic Health Record
One of the reasons hospitals have adopted electronic health record (EHR) systems at such a rapid rate over the past 20 years is the Health Information Technology for Economic and Clinical Health (HITECH) Act of 2009, which provided hospitals and medical practices with $30 billion in incentives to adopt EHR systems [30]. The number of hospitals employing the fundamental EHR system has grown since 2008, according to the Office of the National Coordinator for Health Information Technology. To carry out clinical informatics activities using machine learning, several studies use EHR data. In terms of preprocessing time and feature extraction, the new strategy performs better than the existing approaches. This is brought on by both the vast amount of patient data and the growing use of machine-learning techniques.
2. ARTICLE SEARCHING PROCESS
This section explains the process used for selecting the article for this literature survey.
2.1. Information Sources
We start our research on the target articles by selecting the following online databases:
1. Pub-Med is one of the world's most comprehensive databases in the fields of medicine, life science, bioinformatics, and other related science fields.
2. The Web of Science (WoS) is a large directory that is indexed for multidisciplinary research. The goal of selecting this database was to provide a comprehensive evaluation of scientists' contributions to a field of research, as well as to include important technical papers.
3. The Elsevier’s database contains journals as well as technical and scientific articles.
4. The IEEE Xplore dataset includes scientific papers in electrical engineering, electronics, computer science, bioinformatics, and other related fields.
5. The largest abstract database for peer-reviewed literature is Scopus (i.e., scientific journals and conference proceedings).
2.2. Compilation of Research
Scanning and screening for literature resources are the two stages of the compilation of research. The first stage is to peruse titles and abstracts to eliminate duplicates and useless articles, and the second step is to read the whole article.
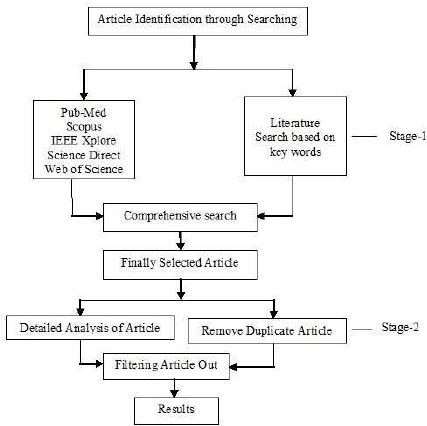
2.3. Search
Early in January 2021, advanced search boxes in the databases of PubMed, WoS, Elsevier’s, IEEE Xplore, and Scopus were used to start this study. To conduct our research, we employed a combination of keywords such as “machine learning,” “bioinspired computing,” “DNA sequence optimization,” and “bioinformatics.” The exact query phrases used in this investigation are shown in Fig. (1), which combines these keywords with the operators “OR” and “AND.” Journal and conference articles were the two types of articles we focused on, and other sorts of reports and book chapters were removed using the search engine's parameters. The two categories were considered to include the most current and relevant scientific journals from our survey.
2.4. Eligibility Criteria
The criteria that each item must fulfill are shown in Fig. (1). The initial objective was to create a wide and coarse-grained taxonomy of nine basic bioinspired algorithms for research on machine learning and bioinspired computing applications in bioinformatics. We use Google Scholar to gain a preliminary understanding of the context and goals of linked papers, and we build the categories based on an unrestricted presurvey of related research. After the initial reduction of duplicates, the remaining articles were excluded by filtering and screening if their eligibility criteria remained insufficient. Exclusion criteria included the objectives of bioinspired computing challenges and solutions to multiobjective optimization issues in bioinformatics. To simplify the further data collection and processing methods, a single Excel file was used that contained a detailed list of all articles from various sites with their corresponding initial classes. As a result of our extensive full-text reading, we have a growing collection of highlights and comments on the surveyed works, as well as a continuing classification of articles into a sophisticated taxonomy. Techniques such as tabulation, description, and summarization were used to reach the main conclusions. Excel and Word files were used to preserve the source databases, their whole list of articles, description tables, summaries, classification tables based on bioinspired algorithms, review sources, objectives, number of features, and approaches used to construct ML, as well as some related data.
2.5. Statistical and Result Information of Articles
The initial search resulted in 2185 articles in the five databases: 75 in PubMed, 1413 in Scopus, 112 in IEEE Xplore, 534 in ScienceDirect, and 51 in WoS. This analysis classified selected papers published between 2003 and 2022 into nine categories (of selected bio-inspired algorithms). The total number of articles from all categories was reduced to 120 after titles and abstracts were skimmed. This number included 29 duplicate articles, and 49 articles were left out of the final full-text reading and evaluation. Given the various topics related to bio-inspired computing challenges and solutions to multi- objective optimization problems in bioinformatics, a total of 91 articles were included in the final set.
3. LITERATURE SURVEY
In this section, we survey some crucial bioinspired computing algorithms applied in bioinformatics with their applications and attempt to present a summary of current advances in the field of bioinspired optimization strategies in bioinformatics. It acts as a torchbearer, encouraging and inspiring more research in all areas of optimization. This survey seeks to provide researchers with the most recent theoretical findings for bioinspired optimization algorithms in the field of bioinformatics.
3.1. Genetic Algorithm and its Application in Bioinformatics
Early in 1962, John Holland's study on adaptable systems established the basis for ensuing research on adaptation in both natural and artificial systems [31]. Then, in 1992, John Koza developed algorithms that performed certain tasks using a genetic algorithm [31]. His process was dubbed “genetic programming” by him. To find accurate or feasible solutions to optimization and search problems, computers can use a type of search strategy called a genetic algorithm. Selection, crossover, and mutation ideas from evolutionary theory are used in genetic algorithms, a kind of evolutionary algorithm. According to a set of rules, people are chosen to have children and contribute to the next generation's population. The selection, which is frequently stochastic, may be influenced by the candidates' test results. By switching chromosomal or genetic segments, two-parent strings are crossed to produce children (new solutions). The probability of crossover is higher, usually between 0.8 and 0.95%. Mutation, on the other hand, is achieved by reversing some of the digits in a string, resulting in the generation of new solutions.
3.1.1. Genetic Algorithm (GA)
Step 1: Initializes the population randomly.
Step 2: Find the fitness of the population.
Step 3: Repeat until the termination criteria are met:
- Choose parents from the general population.
- Crossover results in a new population.
- Create a new population and experiment with mutations.
- Determine a new population's fitness.
Step 4: Return to step 3 until the halting criteria are met.
3.1.2. Literature Survey on GA and its Application in Bioinformatics
This section examines the current innovative genetic algorithms and their applications over the last several decades of computational biology research and development. Iqbal et al. [32] report employing genetic algorithms to orient contentious graph edges to uncover paths in protein-protein interaction networks. Their work reconstructs biologically significant routes in yeast species' weighted networks of protein interactions. The proposed method arranges the edges of the weighted network, and PGMOGA reconstructs paths. They also compare their model with four cutting-edge techniques, and the experimental results perform better. Bader et al. [33] used intelligent algorithms to perform automatic clustering of DNA sequences.
The basic bat algorithm and the popular GA were combined to create the innovative hybrid GABAT algorithm, which was proposed as a solution to the automated data clustering issue. The test findings show that the hybrid algorithm was superior to both the bat algorithm and the fundamental genetic algorithm. To statistically validate the built clusters, the Mann-Whitney-Wilcoxon rank-sum test was performed, and it generated a p-value of less than 5%. Suri et al. [34] provide a framework for objective genetic algorithm optimization using a chaos-DNA hybrid method. In the second step, the suggested model employs two fitness functions with an average changing intensity and entropy with the number of pixel change rates, as well as optimizing the encrypted data. It improves single-objective function optimization significantly. Chaudhary et al. [35] offer a genetic strategy for biological sequence alignment that includes controlled crossover and directed mutation. Their methods are a revolutionary genetic algorithm-based alignment technique that is presented for determining the best alignment of a sequence pair in an efficient manner. They also compared the suggested method's analytical and statistical performance to that of existing comparable sequence alignment approaches. The experimental findings improve the performance of the proposed model by allowing for faster convergence to the optima. Ahmed et al. [36] propose automated dermatological image segmentation by combining a new hybrid intelligent ACO-GA algorithm with a TSVM classifier for disease detection. In dermatological image segmentation for disease diagnosis and their suggested algorithm is used to segment various types of skin lesions. Following image segmentation, their proposed algorithm classified pixels in the image into different classes to investigate precise skin diseases. The hybrid ACO-GA can identify 24 different types of skin diseases. An EPGA-SC is proposed by Lio et al. [37] for the assembly of single-cell sequencing reads. Their contributions include an error-correcting method for nonuniform sequencing, data, reading categorization to a lower percentage of false reads, and the adoption of several sets of high-precision paired-end reads produced from high-precision assemblies. Their experimental findings demonstrate better assembly than the majority of modern tools. Zhang et al. [38] present a model for heart failure disease prediction based on GA to optimize an extreme learning machine. Their model possesses characteristics such as test-training speed and strong generalization ability. Their simulation results have better predictive performance and can better predict the occurrence of heart failure. This model could help doctors predict who will develop heart failure. Finally, Table 2 represents the literature survey on DNA sequencing by using GA application on bioinformatics with advantages and disadvantages.
| Authors/Refs | Year of Publication | Inspiration | Methodology | Advantage | Disadvantage |
|---|---|---|---|---|---|
| Iqbal et al. [32] | 2020 | Evolution Theory | GA | Protocols used in networks of protein- protein interactions |
Only 20% of the tests had improved outcomes. |
| Bader et al. [33] | 2021 | Evolution Theory, BAT | GABAT | Comparing sequence similarity analysis | Sequential information is not found individually. |
| Suri et al. [34] | 2017 | Evolution Theory | GA | It improves performance of encryption | Complex to perform and manage this algorithm |
| Chaudhary et al. [35] | 2014 | Evolution Theory | GA | An equal number of chromosomes are swapped from one parent to another and vice versa | It works on a smaller DNA sequence pair |
| Ahmed et al. [36] | 2019 | Evolution Theory, ACO | ACO-GA | Skin disease detection accuracy is 95%. | Does not include large dermatological images for skin disease |
| Lio et al. [37] | 2019 | Evolution Theory | GA | Generates better sequence assemblies | The running time is high and takes a large amount of memory |
| Zhang et al. [38] | 2021 | Evolution Theory | GA | It produces better prediction results. | It is not an advanced model for disease prediction. |
3.2. PSO and its Application in Bioinformatics
Based on the social behavior of animal groups such as insects, cattle, birds, and fish, Eberhart, and Kennedy proposed the particle swarm optimization (PSO) algorithm as a stochastic optimization technique in 1995 [39]. Each swarm member improves the search pattern depending on its and other members' learning experiences, which can assist the swarm. While a bird searching for food at random can improve its search by working with groups and their cooperative approach to finding food. The location and velocity of each particle are updated as follows [40, 41]:
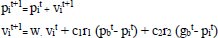
Where pit represents Particle position.
vit represents particle velocity.
Cognitive and social factors are represented by c1 and c2
r1 r2 are arbitrary values between 0 and 1.
3.2.1. Particle Swarm Optimization Algorithm (PSO)
Step 1: Construct a population of particles that are distributed uniformly along a vector X.
Step 2: Use the objective function to determine each particle's position.
Step 3: If a particle's current location exceeds its previous top position, its velocity should be updated.
Step 4: Select the best particle (according to the particle's last best position).
Step 5: The velocity of the particle is updated:
vit+1= w. vit + c1r1 (pbt- pit) + c2r2 (gbt- pit)
(pbt -Local best solution, gbt-Global best solution)
Step 6: Distribute the particles to their new position:
pit+1= pit+ vit+1
Step 7: Continue with step 2 until the termination criteria are met.
3.2.2. PSO Literature Survey and its Application in Bioinformatics
This section examines the current innovative PSOs and their applications over the last several decades of computational biology research and development. Papetti et al. [42] apply particle swarm optimization techniques to simplify global optimization. For a multidimensional search space, they propose a dilation function. The search space can improve the fitness distribution by creating a random population and improving the overall optimization process. This automated approach will allow us to investigate the performance of local bubble dilation functions on challenging optimization issues in real-life scenarios. Zhu et al. [43] provide a novel approach for better PSO based on elastic collisions for DNA coding design. Using the sparrow search algorithm, the suggested EC-PSO enhances the optimal and worst positions within the population. The harmony search technique is then introduced to increase the quality of DNA sequencing. The algorithm's effectiveness is validated by eight test functions. In the DNA sequence design, their model was more rational, and the produced sequences were of greater quality. Som-In et al. [44] present an improved particle swarm optimization-based approach for detecting different motifs in DNA sequence collection. They proposed PSO-HD, which improves the efficiency of finding transcription factor binding sites (TFBS) by using particle swarm optimization with Hamming distance (PSO-HD). They compare the related algorithms using the F1-score as a measurement unit. The experimental results increase the detection of TFBS. Khan et al. [45] proposed a modified PSO technique for solving DNA problems. To evaluate multiobjective issues in DNA sequencing, the proposed technique makes use of four objective functions. To determine how effective the proposed method is, they use average and standard deviation data. Results of the experiment show that the recommended technique outperforms others. Vijayalakshmi et al. [46] applied particle swarm optimization with non-dominant sorting to give a multimodal prediction of breast cancer. They proposed a multimodel prediction strategy for breast cancer. The model makes use of publicly available WBCD and WDBC ultrasonic computed tomography breast cancer data sets (UCI). The results of the findings showed that kernel density estimation performed well (98.8% and 98.3%), which is extremely promising. Indumathy et al. [47] proposed a model using PSO with inertia weight and constriction factor to solve DNA sequence assembly problems. The proposed strategy optimizes the DNA fragments' overlapping scores. To find the optimal answer, they use computational tools. According to the experimental data, the proposed method beats earlier techniques in terms of overlap score. Elsayed et al. present a new hybridization technique for recreating DNA sequences based on probabilistic cellular automata with PSO [48]. The proposed model is split into two parts. The first phase involves analyzing the evolution of DNA sequences, and the second involves reproducing the sequences. Using cellular automata models, the DNA sequence is then analyzed and predicted. In this article, they assess a large number of DNA sequences to anticipate the changes that occur in DNA mutations. Wang et al. [49] present a PSO-based approach for generating DNA barcode sets. In this work, a PSO technique was adapted and used to build DNA bar code sets that use GC content as ideal self -complementary and continuity constraints. The GC content ranges between 40% and 60%. The proposed PSO method is used to generate new DNA barcode sets to improve lower limits, and the results are essentially equivalent to or better than the modified genetic algorithm that improves the lower bounds of DNA barcode sets. Using PSO and an ensemble learning approach, Wang et al. [50] created a feature fusion predictor for RNA pseudouridine sites. They presented PsoEL-PseU, a new feature fusion predictor for pseudouridine site prediction. They use binary PSO to find the best feature subset for each of the six feature subsets. Using the six best feature subsets, they trained six individual predictors. They also used a sliding window technique to get around the predictors' inability to identify RNA sequences inside nonterminal lengths. In addition, powerful deep-learning techniques are used to determine RNA sequence information. Finally, they demonstrate that the improved predictor performed better in the independent dataset evaluation. Using coupled BA-PSO and infection propagation, Issa et al. [51] propose a technique for detecting biological subsequences in COVID-19. This article proposes a modified version of FLAT that uses the hybridization of the PSO method with the BA algorithm to improve the performance of the BA algorithm. By using BA operators to find the optimum solution, the recommended technique, BPINF, identifies the best-investigated solution. The proposed model outperforms all existing state-of-the-art models in detecting LCCSS between a set of real biological sequences with substantial lengths, including COVID-19 and other well-known viruses, with an accuracy of 88 percent. Goncalves et al. [23] present CNN architecture optimization using bioinspired cancer detection in infrared images. The goal of the suggested paper is to demonstrate that early identification of breast cancer is critical to improving patient cure and recovery rates. In this paper, they apply bioinspired optimization techniques to determine suitable hyper-parameters and the architecture of completely connected layers. They were also able to improve the layer's ResNet-50 outcome from 0.83% to 0.90% of the F1 scores. Liu et al. [53] present a BPSON algorithm that has been applied to the construction of DNA codes. The bat algorithm and the PSO algorithm are combined in this BPSON algorithm. To defeat PSO and improve the global search ability, a bat algorithm is utilized in this article. The experimental results reveal that the suggested approach may avoid the formation of secondary structures as well as hybridize them. The results demonstrate that their technique may generate an optimal code for DNA computing, and the performance is compared with the existing model. Table 3 depicts the literature survey on DNA sequencing by using PSO application in bioinformatics with advantages and disadvantages.
Table 3.
| Authors/Refs | Year of Publication | Inspiration | Methodology | Advantage | Disadvantage |
|---|---|---|---|---|---|
| Papetti et al. [42] | 2022 | Local Bubble | PSO | Dimension of the search space is efficient | Can’t use periodic boundary function |
| Zhu et al. [43] | 2022 | Sparrow, harmony | PSO | Determine the higher-quality DNA sequence | Computation of DNA sequence is a complex process |
| Som-In et al. [44] | 2020 | Birds | PSO | Improve computational time | It can’t detect TFBS from various datasets |
| Khan et al. [45] | 2019 | Birds | MPSO | Creates a single objective function out of a multiobjective issue. | Difficult to perform large datasets |
| Vijayalakshmi et al. [46] | 2020 | Birds | PSO | Identify patterns of breast cancer cells in groups of samples | Evaluations of the quality of data are a complex process |
| Indumathy et al. [47] | 2014 | Birds | CPSO | It maximizes the overlapping score of DNA fragments | Complexity while computing a lengthy sequence. |
| Elsayed et al. [48] | 2021 | Birds | Cellular automata PSO | Predicting DNA structural changes during mutations | Difficult to perform large dataset |
| Wang et al. [49] | 2018 | Birds | PSO | Compared to other algorithms, it increases the prediction accuracy. | This algorithm is difficult to execute and run. |
| Wang et al. [50] | 2021 | Birds | BPSO | It gets better accuracy in the independent data set evaluation | More computational resources consumed with a new species |
| Issa et al. [51] | 2022 | Birds, Bat | BA-PSO | Predict disease by Bio-inspired machine learning algorithm. | The complexity of computation with a long sequence |
| Goncalves et al. [52] | 2022 | Evolution Theory, Birds | GA-PSO | Prediction of diseases like cancer | A collision occurs while using a large dataset. |
| Liu et al. [53] | 2019 | Birds | PSO | Sort the DNA sequences | It is difficult to calculate the 'cc' pair in the genome sequence. |
3.3. ACO and its Application in Bioinformatics
Early in the 1990s, Macro Dorigo and colleagues [54] introduced the first ACO algorithm in their Ph.D. thesis. Real ant colonies were used to construct this algorithm. Ant colony optimization (ACO) is a meta-heuristics based on the group of ant colonies. The core concepts of ACO are self-organizing principles, which illustrate how artificial ant populations may cooperate to solve complex problems by displaying coordinated behaviors similar to those of real ants. Essential components of the ACO's optimization problem are the state transition rule and the pheromone update rule [16]. The initial vertex, a probabilistic technique, is used when an ant chooses which vertex to visit next. The second one selects previously traveled edges to a greater extent. Each edge of graph G of visiting cities i to j has a pheromone trail τij connected to it.
Where,
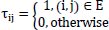
The pheromone evaporation is carried out as follows once each ant has finished its turn:
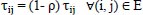
Where ρ ϵ (0, 1) is a parameter that simulates the rate at which the pheromone intensity evaporates. The heuristic function, ηij= 1⁄dij is used to assess the quality of items that can be included in the current partial solution. The arc has the highest probability
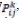 , which belongs to a particular arc-related vertex. The next edge of graph G is chosen based on the state transition probability of ant k, which is described as follows. An ant will travel from node i to node j based on probabilities [28].
, which belongs to a particular arc-related vertex. The next edge of graph G is chosen based on the state transition probability of ant k, which is described as follows. An ant will travel from node i to node j based on probabilities [28].
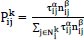
If an ant named K is in city i, then
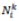 is a possible neighbourhood for them. According to two parameters, the pheromone trails and the heuristic information's respective effects are determined [54]. This section defines the function of parameters α and β [34]. The probability of selecting a city increases if α=0.If β = 0, no heuristic bias is present, and simply pheromone amplification or the application of pheromone is being employed. This generates relatively poor outcomes, and, in particular, for values of α > 1, it quickly generates a path of stagnation problem [55].
is a possible neighbourhood for them. According to two parameters, the pheromone trails and the heuristic information's respective effects are determined [54]. This section defines the function of parameters α and β [34]. The probability of selecting a city increases if α=0.If β = 0, no heuristic bias is present, and simply pheromone amplification or the application of pheromone is being employed. This generates relatively poor outcomes, and, in particular, for values of α > 1, it quickly generates a path of stagnation problem [55].
3.3.1. ACO Algorithm
Step 1: Each ant's journey represents a potential solution to a particular problem.
Step 2: The amount of pheromones left behind on a path by an ant corresponds to the problem's quality.
Step 3: The path with the highest pheromone has a higher probability of being selected when an ant must choose between two or more paths.
Step 3.1 Once all ants have completed their journey to meet the requirement; we continue to apply the pheromone updating rule.
Step 4: Produce the best worldwide solution.
3.3.2. Literature Survey on ACO and its Application in Bioinformatics
This section examines the current innovative Ant Colony Optimization algorithms and their applications over the last several decades of computational biology research and development. A novel hybrid ant colony optimization method was developed by Bir-Jmel et al. [56] for the classification of cancer in high dimensional data. This algorithm uses a model of gene selection. Based on adopting a new graph-based technique for gene selection, they propose a hybrid strategy (MWIS-ACO-LS) for the gene selection problem in their work, to minimize gene redundancy. They put their approach to the test on ten high-dimensional microarray datasets ranging from 2308 to 12600 genes. Their suggested technique is effective, as shown by the experimental findings. To solve the DNA sequence design problem, Misinem et al. [57] developed a new hierarchical pheromone update system and population-based ant colony optimization. They describe an improved multiobjective optimization approach called PACO. To construct the DNA sequence, they employ a finite state machine model. Each level uses a 20-level pheromone matrix. Each node can connect to the next node in the level via four different paths. Each pheromone matrix has a mechanism for exchanging its information with another pheromone matrix, allowing them to access the best results from other populations. The findings indicate that this new approach can produce a more exact DNA sequence. To optimize DNA sequences, Kurniawn et al. [58] created an ant colony system. To deal with the DNA sequence optimization challenge, in this work, the ant system (AS) was introduced. The method used to find solutions based on pheromone data involves using certain ants. Four nodes—which stand for the DNA bases—make up its structure. The results of the proposed strategies are contrasted with those of alternative techniques, such as the evolutionary algorithm. Wang et al. [59] proposed an intelligent privacy-preserving technique for discovering associations in genomic association studies. They devise a multiobjective search strategy to find prospective single-nucleotide polymorphism (SNP) sets linked to an illness phenotype to develop a logical epistemic privacy protection technique. They employ four widely used techniques to find K-order SNPs. The experimental results indicate that detecting numerous models improves search accuracy and makes the framework more stable. For DNA sequencing by hybridization, Blum et al. [60] propose an ant colony optimization technique. To determine the average global similarity scores for their research, they provide a multilevel framework that uses the ant colony optimization technique for DNA sequencing. The results suggest that the ML-ACO techniques have performed better than conventional techniques. Sun et al. [61] improve the ant colony optimization approach for locating epistasis with heuristic data, which is then incorporated into ant decision rules in the linear time. Their recommended techniques have an O(Lj + nm2) temporal complexity, where L is the number of ants, j is the number of iterations, n is the number of SNPs, and m is the number of samples. It requires less time to implement the proposed method in practical applications. To find evolutionary relationships between species that are closely related based on their distance, Perera et al. [62] proposed a modified ant colony-based algorithm for phylogenetic tree construction. In regard to creating distance matrices and calculating Kolmogorov complexity as a measure of distance between species from an entire mitochondrial genome alignment, their recommended method performs better than conventional techniques. The summary of the literature survey on DNA sequencing by using ACO application on bioinformatics with advantages and disadvantages is represented in Table 4.
3.4. Bat Search Optimization and its Application in Bioinformatics
Bats are the only intelligent creatures with wings who can detect prey via biosonar. These bats emit an extremely loud sound pulse and listen for the echo that is reflected from nearby objects (prey). For echolocation, the majority of bats rely on constant-frequency transmission. Depending on the species, their pulses have different features and can be linked to hunting techniques. Yang published the bat in 2010 which spurred the development of this metaheuristic method [63]. Yang published a new Multi-objective bat search algorithm in 2011. The criteria for updating their locations and velocities in an n-dimensional search space are described. New solutions and velocities are generated at time t as follows:
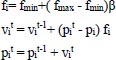
In this case, β ϵ [0,1] is a random vector selected at random from a uniform distribution. Here is the current global best solution (position) across all “m” bats.
3.4.1. Bat Search Algorithm (BS)
Step 1: Initialize the bat population, including all bats' positions, velocities, and frequencies.
Step 2: Set all bats' echolocation parameters.
Step 3: After evaluating the bats in their initial position, the determined answer is saved.
Step 4: In descending order, sort the current population.
Step 5: Produce candidate bat solutions.
Step 6: Fly at random by changing your position, velocity, and direction.
Step 7: Select a bat at random from the population.
Step 8: Assess the bat based on its fitness function.
Step 9: Update the echolocation parameters for a more accurate next position.
Step 10: Choose the best bat for the next iteration.
Step 11: Repeat Steps 4 through 10 until the termination criteria are met.
3.4.2. Literature Survey on BS and its Application in Bioinformatics
This section examines the current innovative Bat Search Optimization algorithms and their applications over the last several decades of computational biology research and development. Zemli et al. [64] describe a model that solves numerous sequence alignment challenges by using a bioinspired approach. They introduce a new bioinspired approach to solving problems by employing BA-MSA (Bat Algorithm-Multiple Sequence Alignment). This strategy offers a new mechanism for generating the initial population.
Table 4.
| Authors/Refs | Year of Publication | Inspiration | Methodology | Advantage | Disadvantage |
|---|---|---|---|---|---|
| Bir-Jmel et al. [56] | 2019 | Ants | ACO | The most effective way to choose a subset of genes for cancer identification | From a dataset, it selects a limited handful of critical genes. |
| Misinem et al. [57] | 2019 | Ants | PACO | It generates better DNA sequences | Difficult to perform large data set |
| Kurniawn et al. [58] | 2009 | Ants | ACO | The DNA sequence is better than other existing approaches | The generated sequence has less continuity observed |
| Wang et al. [59] | 2022 | Ants | ACO | Machine learning-based disease prediction | the difficulty of computing with a large sequence |
| Blum et al. [60] | 2008 | Ants | ACO | Justified sequence similarity analysis | The complexity is higher |
| Sun et al. [61] | 2018 | Ants | ACO | It takes linear time. | Inadequate heuristic data to optimize the search |
| Perera et al. [62] | 2019 | Ants | ACO | It takes less processing time | The dataset is short |
| Authors/Refs | Year of Publication | Inspiration | Methodology | Advantage | Disadvantage |
|---|---|---|---|---|---|
| Zemli et al. [64] | 2016 | Bat | BA-MSA | It improves accuracy in the prediction of multiple sequence alignment | Complex to perform and manage this algorithm |
| Al-Betar et al. [65] | 2020 | Bat | MBA | Predict defective genes by the machine learning algorithm | It does not suit high-dimensional datasets. |
| Kaur et al. [66] | 2021 | Bat | PF-BAT | It provides a fast automated intelligent tool for decision making | Collision occurs while using a large dataset. |
| Badr et al. [33] | 2021 | Bat | Clustering-BAT | Intelligent Techniques for Clustering | Less effective |
It entails creating a guide tree for each solution using a progressive method and altering some parameters. For experiments, they employ the Balibse 2.0 dataset. They used a statistical non-parametric test to determine whether BA-MSA increases the effectiveness of the proposed strategy. Al-Betar et al. offer a bat algorithm inspired by Triz [65] for gene selection in cancer categorization. A hybrid rMRMR-MBA for the gene selection problem is presented in this paper. To find the most promising genes in this model, the robust minimum redundancy maximum relevance (rMRMR) is used as a filter and the modified bat algorithm (MBA) is used as a search engine to select a small number of beneficial genes. Using ten gene expression datasets, the effectiveness of the suggested technique was evaluated. Their proposed strategy is capable of producing highly promising results for gene selection with high classification accuracy. Kaur et al. [66] introduced the automated diagnosis of COVID-19 by using deep features and parameter-free bat optimization. In this work, deep learning models are trained to assess the severity of COVID-19 using CT scans. Researchers have developed a parameter-free bat (PF-Bat) optimized fuzzy k-nearest neighbor (PF-FKNN) classifier to identify the coronavirus. Mobile-fully Net2's connected layer of transfer was used to extract features, and FKNN was used to train the features. The experimental results show 99.38 percent accuracy, which is higher than the previous model and is, based on Covid CT scan benchmark data. Professionals using this model will be able to effectively manage patients' healthcare needs, resulting to a faster recovery. Based on the bat algorithm, Badr et al. [33] introduced an intelligent method for the automatic grouping of DNA sequences. For problems involving automatic clustering, they propose a soft computing based metaheuristic framework. The results of the tests show that their hybrid technique outperformed the bat algorithm as well as the standard genetic algorithm. To statistically validate the development clusters, a Mann-Whitney-Wilcoxon rank-sum test was used. The Wilcoxon test has a p-value of less than 5%. Table 5 depicts the summary of the literature survey on DNA sequencing by using BS application on bioinformatics with advantages and disadvantages.
3.5. Evolutionary Algorithm and its Application in Bioinformatics
The evolutionary algorithm (EA), a subset of evolutionary computing and a generic population-based metaheuristic optimization approach, was developed by 'Back' in 1996 [67]. It makes use of techniques that include reproduction, mutation, recombination, and selection that were inspired by biological evolution. Evolutionary algorithms used to mimic biological approximation solutions to a wide range of problems are frequently limited to assessments of microevolutionary processes and planning models since they ideally make no assumptions about the underlying evolution based on physical processes. An EA employs four procedures inspired by evolutionary biology: initialization, genetic operator, selection, and termination:
3.5.1. Initialization
Creating a fitness function can be tricky, so it's critical to select the one that appropriately represents the facts in the problem. Then, after calculating the fitness of all members, pick a subset of the highest-scoring members.
3.5.2. Genetic Operator
Crossover has a higher chance of producing new solutions by swapping genes. First, we must generate a population of solutions. The population will have an arbitrary number of members, which are viable solutions to the problem.
3.5.3. Selection
After a population has been generated, members of the population will be evaluated based on their chromosomes. When a mutation is performed by flipping some of a string's digits, new solutions are produced, and the chance of mutation is quite low.
3.5.4. Termination
When an algorithm reaches its finish, one of two things normally happens: either the program has reached its maximum runtime or it has reached a performance threshold. After that, a final solution is chosen and returned.
3.5.1.1. Evolutionary Algorithm (EA)
Step 1: Create a population of potentially good solutions by selecting them at random.
Step 2: Calculate each person's fitness in the group.
Step 3: Use a selection technique to pick the parents.
Step 4: Crossover and mutation operators can be used to generate offspring.
Step 5: Assess the new offspring's fitness.
Step 6: Using a selection technique, pick members of the population to die.
Step 7: Repeat Steps 2–7 until all termination conditions have been met.
3.5.1.2. Literature Survey on EA and its Application in Bioinformatics
This section examines the current innovative evolutionary algorithms and their applications over the last several decades of computational biology research and development. Eremeev et al. [68] presented a model for estimates based on evolutionary algorithm theory applied to gene design. They use a SELEX approach to locate a group of motifs that meet the target area of the genotypic space by placing an upper limit on the expected hitting time. The application of EA on expected fractions of DNA sequences that are properly matched during a particular iteration. Their experimental results imply some theoretically provable bounds for EA performance. A multiobjective evolutionary method is used by Lee et al. [69] to optimize DNA sequences. They use a restricted, elitist, non-dominating genetic algorithm with a random selection. It is the first time a multiobjective and limited methodology has been used to get the best possible collection of DNA sequences. They evaluated the performance by comparing it to a previously published DNA sequence. A model for creating DNA sequences based on a multiobjective memetic generalized differential evolution technique was presented by Bano et al. [70]. In this study, the population-based memetic generalized differential evolution (MGDE3) method is used to meet the following four design goals for reliable DNA sequence design. They evaluate their algorithm against seven well-known multiobjective methods using small, medium, and large data sets. The proposed technique MGDE3 outperforms the compared algorithms in both experimental and statistical analysis. It produces a more precise DNA sequence in less amount of time than existing models. A literature survey on DNA sequencing by using EA application on bioinformatics with advantages and disadvantages represents in Table 6.
3.6. Cuckoo Search Optimization and its Application in Bioinformatics
Yang and Deb [71] proposed the Cuckoo Search metaheuristic optimization technique in 2009. The invention of a novel optimization method was sparked by the laying and breeding of cuckoo eggs. It is a metaheuristic optimization approach with natural inspiration for resolving optimization issues. With this approach, NP-hard combinatorial optimization tasks are more effective, accurate, and likely to converge. Because the cuckoo can only lay one egg at a time, each egg represents a solution that is kept in the nest. The cuckoo nest ‘i’ adjusts its position in each iteration, and the new generating solution for iteration is as follows:
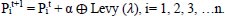
α = step size
λ = Levy exponent
⨁ = Entry-wise multiplication
Pit = position of the nest
3.6.1. Cuckoo Search Algorithm (CS)
Step 1: Create an N-host nest population, with each h1 and c1 being a candidate for optimal parameters.
Step 2: Every time a cuckoo lays an egg, they choose a nest at random to place it in.
Step 3: Compare the cuckoo egg's fitness to the host egg's fitness.
Step 4: If the fitness of the cuckoo's egg exceeds that of the host egg, the cuckoo's egg will be replaced with another host
nest.
Step 5: If the host bird notices it, it can either discard the egg outside the nest or abandon it and establish a new one (nest).
Step 6: Repetition of Steps 2–5 until the termination criteria are met.
Table 6.
| Authors/Refs | Year of publication | Inspiration | Methodology | Advantage | Disadvantage |
|---|---|---|---|---|---|
| Eremeev et al. [68] | 2018 | Evolution Theory | EA | It identifies DNA or RNA sequences with high affinity | It does not compare their result with existing methods |
| Lee et al. [69] | 2003 | Evolution Theory | EA | It generates optimized DNA sequence | Computation of DNA sequence is a complex process |
| Bano et al. [70] | 2022 | Evolution Theory | EA | It generates better DNA sequence | Difficult to perform larger dataset |
| Authors/Refs | Year of publication | Inspiration | Methodology | Advantage | Disadvantage |
|---|---|---|---|---|---|
| Indumathy et al. [72] | 2015 | Cuckoo | CS | Compare the common pattern of the sequence | Difficult to perform large dataset |
| Othman et al. [73] | 2020 | Cuckoo | CS with evolutionary operators | Identifying cancer genes | Minimize the number of genes and maximize the relevance of the selected genes |
| Alyassri et al. [74] | 2022 | Cuckoo | MOBCS-KNN | It improves the accuracy rate of EEG channels | Its performance is not accurate while selecting fewer channels. |
| Yin et al. [75] | 2018 | Evolution Theory, Cuckoo |
GACSB | It improves the quality index of bi-clusters | Evaluation of the quality of data is a complex process. |
| Elewa et al. [76] | 2014 | Cuckoo | MACS | It finds motifs faster than other existing methods | A collision occurs while using a larger dataset |
3.7. Literature Survey on CS and its Application in Bioinformatics
This section examines the current innovative Cuckoo Search algorithms and their applications over the last several decades of computational biology research and development. Indumathy et al. describe a naturally inspired cuckoo search strategy for genomic sequence assembly [72]. It is the first time that CS has been used to tackle a DNA sequence assembly problem. It is used to maximize the overlap score of the original DNA sequence. In terms of high-quality solutions, CS trumps PSO and its variants. Othman et al. [73] suggested a cuckoo search algorithm based on bioinspired principles that prioritize key genes over other genes. As a feature selection strategy, their MOCS-EO algorithm intends to apply a multi-purpose approach that minimizes the number of genes while increasing the relevance of selected genes and cancer classes. This algorithm chooses a small number of genes for decision-making, resulting in higher classification performance than using all genes that vary in all seven minor parameter settings of the cancer microarray dataset. The suggested Multi-objective cuckoo search with evolutionary operators outperforms cuckoo search and multi-objective cuckoo search algorithms with a small number of key genes.
As a safety measure in a healthcare application, Alyassri et al. [74] describe EEG channel selection for identification systems using a multiobjective cuckoo search. Instead of balancing the accuracy and the total number of selected EEG channels in their study, they focused on improving the accuracy rate. The proposed model is a weighted sum-based multi-objective binary cuckoo search algorithm (MOBCS-KNN) that constructs a multiobjective approach. The trials' findings demonstrate that the suggested model achieved 93.86 percent accuracy with just 20 sensors and AR20 auto-regressive coefficients. It removes redundant and undesired features while maintaining specific features. Yin et al. [75] demonstrate biclustering of gene expression data using cuckoo search and a genetic algorithm. Gene expression data analysis in their model can uncover a large number of biologically meaningful local gene expression patterns. To answer the problem of gene expression data clustering, they presented a metaheuristic approach based on GA and CS algorithms (GA-CS biclustering, Georgia Association data of community service Boards) GACSB in this article. The results show that GACSB enhances the quality index and coverage of biclusters when compared to other existing algorithms. Elewa et al. [76] propose a variant of the cuckoo search algorithm for motif-finding problems. In this study, they handle plated motif problems using the CS approach. In comparison with other existing algorithms, the experimental results indicate that the suggested MACS adaptation can locate motifs fast and efficiently. Table 7 depicts the summary of the literature survey on DNA sequencing by using CS application in bioinformatics with advantages and disadvantages.
3.8. Grey Wolf Optimization and its Application in Bioinformatics
In 2014, Mirjalili et al. [77] introduced the Grey Wolf Optimization approach as a brand-new metaheuristic optimization method. It is a population-based stochastic method for selecting the most effective solution from a list of solutions (population). This algorithm is based on the social hunting behavior of grey wolves, which involves three steps: tracking, encircling, and attacking. The algorithm optimizes by mathematically modeling how grey wolf populations track, surround, hunt, and attack. Based on the location of the prey, the aforementioned equations are employed to modify the grey wolf's position.
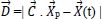
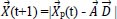
Where
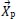 is the position vector of the prey,
is the position vector of the prey,
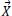 is the position vector of a grey wolf, t is the current iteration,
is the position vector of a grey wolf, t is the current iteration,
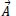 and
and
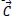 are coefficient vectors. The following formula is used to calculate the vectors
and
:
are coefficient vectors. The following formula is used to calculate the vectors
and
:
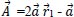
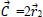
Where r1 and r2 are random vectors in the range [0, 1] and components of
 are linearly decreasing from 2 to 0 during the time of iteration cycles.
are linearly decreasing from 2 to 0 during the time of iteration cycles.
3.8.1. Grey Wolf Optimization Algorithm (GWO)
Step 1: Set the population size and maximum iteration settings.
Step 2: Evaluate all search agent's fitness.
Xα = most effective first searching parameter of the agent.
xβ = second searching agent’s parameter.
xδ = most effective third searching agent’s parameter.
Step 3: Once the given number of iterations has been achieved, evaluate each agent's fitness.
Step 4: Next, the position of the current search agent is updated, and the fitness level of each search agent is evaluated.
Step 5: Repeat Steps 2–5 until the termination requirements have been met.
Step 6: Generates the best solution xα.
3.8.2. Literature Survey on GWO and its Application in Bioinformatics
This section examines the current innovative GWO algorithms and their applications over the last several decades of computational biology research and development. Nematzadeh et al. [78] describe how to tune the hyperparameters of machine learning algorithms and deep neural networks using metaheuristic applications in biomedical and biological cases.
They introduce the tuning hyperparameters of machine learning algorithms using Grey Wolf optimization and the evolutionary algorithm. Their proposed method employs 11 distinct algorithms and datasets in biological and biomedical domains to demonstrate and experimental results deliver greater performance with faster convergence, and the optimizer aims to decrease the algorithm cost (space/time complexity). Using the Grey Wolf optimizer and operators with TRIZ-inspired design, Alomari et al. [79] provided a model for gene selection for microarray data classification. The production of a single chip with 1,000 genetic instructions using DNA microarray technology is well recognized. By using nine well-known microarray datasets and their proposed modified Gray-wolf optimizer, which evaluates the quality of a selected group of genes using support vector machines for classification tasks, they can identify the top-ranked genes. Additionally, they contrasted the results of their study with seven cutting-edge gene selection techniques. Six of the nine datasets were able to pinpoint the ideal gene combination, and their results are the best in four of the dataset. Chen et al. [80] provide a model for grey wolf optimizer adaptation for the disassembly sequencing problem. They investigate a grey wolf optimizer (GWO) for continuous optimization issues in their study; however, it cannot be immediately applied to a disassembly sequencing problem. The experimental results reveal that SGWO outperforms previous methods. Hasim et al. [81] presented methods for grey wolf optimization for motif discovery (GWOMF). They presented and assessed six synthetic datasets in their research. The experimental findings demonstrate an outstanding success rate with a less processing time that is comparable to other available approaches. With an average success rate of 86.6%, these novel models indicate their effectiveness as a motif discovery tool. Table 8 shows the summary of the literature survey on DNA sequencing by using GWO application on bioinformatics with advantages and disadvantages.
3.9. Whale Optimization Algorithm and its Application in Bioinformatics
The Whale Optimization Algorithm (WOA) is a meta-heuristic optimization technique that takes its cues from nature and mimics the hunting behavior of humpback whales. The bubble-net hunting method served as an inspiration for the algorithm. The bubble-net feeding technique, which Seyedali and Andrew first introduced in 2016, is known as the foraging behavior of humpback whales [82]. Hunting krill or small fish in groups near the surface is preferred by humpback whales. The humpback whale can locate and encircle prey. The WOA approach assumes that the target prey or a close relative of it is the best candidate solution at this time because the location of the optimum design in the search space is unknown in advance. The other search agents will make an effort to update their positions to that of the best search agent when the best search agent has been identified. These equations describe the whale's behavior:
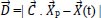
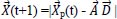
Where
denote the position vector of the hunted animal,
denote the position vector of a Whale, t denote the current iteration,
and
denote vectors of coefficients. The following equation is employed to determine the vectors
and
:
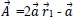
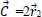
Where r1 and r2 are random vectors in the range [0, 1] and components of
are linearly decreasing from 2 to 0 during the time of iteration cycles.
| Authors/Refs | Year of publication | Inspiration | Methodology | Advantage | Disadvantage |
|---|---|---|---|---|---|
| Nematzadeh et al. [78] | 2022 | Wolf | GWO | Predict new incoming samples accurately and minimize the complexity | Evaluation of the quality of the sample is a complex process. |
| Alomari et al. [79] | 2021 | wolf | Gray-wolf optimizer | Effective outcomes in the gene selection problem | Protein tertiary structure is not predictable |
| Chen et al. [80] | 2019 | wolf | SGWO | It is the first model to solve the disassembly sequencing problem. | Complexity is higher when computed with a large sequence. |
| Hashim et al. [81] | 2017 | wolf | GWOMF | It is the first method for finding motifs using GWO | Difficult to perform large datasets and its computation time is more |
3.9.1. Whale Optimization Algorithm (WOA)
Step 1: Whales’ population is initialized randomly.
Step 2: Then initialize the iteration counter t.
Step 3: Next, let’s initialize the value of a=2.
Step 4: Afterward evaluate each search agent
i by calculating its fitness function.
Step 5: Update the position vector according to
and
.
Step 6: Repeat steps 4 and 5 until the termination requirements have been met.
Step 7: Finally return the best solution.
3.9.2. Literature Survey on WOA and its Application in Bioinformatics
This section examines the current innovative WOA algorithms and their applications over the last several decades of computational biology research and development. A triplet-base unpaired constraint is a novel constraint proposed by Li et al. [83], which can form a new combination constraint by combining it with existing constraints. The Harmony Search (HS) method and the Whale Optimization Algorithm (WOA) are combined to form a novel method known as HSWOA, which they used to produce DNA sequences that adhere to the new combination requirement. Finally, when compared to past findings, their findings show that their approach not only increases the efficiency of hybridization reactions but also generates a greater fitness value. A novel evaluation function, FCC, is presented by Abdel-Basset et al. [84] to assess the quality of various assemblers. In 30 benchmark instances, DWOA-LS was validated in two tests, and it was contrasted against several modern, highly resilient state-of-the-art DFAP algorithms. To show DWOA's superiority in converting continuous whale behaviors to discrete, it was contrasted against the WOA, DE, and SCA versions in the first trial. To show the relevance of the DWOA over those algorithms, the Wilcoxon rank sum test was also run. The purpose of the second experiment was to show that DWOA-LS outperformed some contemporary, reliable, state-of-the-art assemblers that were suggested for the DFAP.
A drawback of the suggested method, in addition to its temporal complexity, is that it occasionally fails to produce better overlap scores than CSA-P2M*Fit. An improved method for selective opposition whale optimization is provided by Wang et al. [85]. The first step is to update the predator's position selectively using improved quasi-oppositional learning (EQOBL), compute the population's fitness before and after, and maintain the positions of the best people as food sources. The position update of the food supply is finished by the second technique, which is an improved time-varying update technique for predator inertia weight position. 23 benchmark functions from CEC 2005 and 15 benchmark functions from CEC 2015 are used to compare the algorithm's performance in several dimensions. The nonparametric rank test by Friedman and the rank sum test by Wilcoxon both provide better outcomes. Applications in the field of biological computing, in conclusion, demonstrate its relevance. Building high-quantity DNA code sets is one of ISOWOA's objectives in their study. The experimental results demonstrate that the lower bounds of the multi-constraint storage coding sets used in this study are on par with or better than those of earlier optimal implementations. The results show that ISOWOA enhanced the quantity of DNA storage codes filtered by 2–18%, proving the algorithm's dependability in real-world optimization tasks. Stephan et al.'s [86] algorithm for a hybrid artificial bee colony (HAW) suggests an employee bee’s attacking phase by combining ABC's exploitative employee bee phase with the whale optimization's bubble net attacking technique. During the attack phase, worker bees employ humpback whales to find better food sources. The proposed mutative initiation step, which is part of the HAW algorithm's exploratory portion, improves on regular ABC's mediocre exploration. The simultaneous feature selection and parameter optimization of an ANN model use the HAW method. Back-propagation learning is used to implement HAW, which consists of momentum-based gradient descent, Levenberg-Marquart, and robust back-propagation (HAW-RP and HAW-GD). Several breast cancer datasets are used to evaluate the accuracy, complexity, and processing time of these hybrid versions. The HAW-RP variant achieved good accuracy using a low-complexity ANN model in comparison to HAW-LM and HAW-GD. Table 9 shows the summary of the literature survey on DNA sequencing by using the WOA application on bioinformatics with advantages and disadvantages.
| Authors | Year of publication | Inspiration | Methodology | Advantage | Disadvantage |
|---|---|---|---|---|---|
| Li et al. [83] | 2020 | Humpback whales | WOA | Reduce the values of the H-measure and the similarity of the DNA sequence set. |
Evaluation of the quality of the sample is a complex process. |
| Abdel-Basset et al. [84] | 2020 | Whales | WOA | Effective outcomes in the DNA Fragment Assembly Problem |
It does not achieve better overlap scores |
| Wang et al. [85] | 2022 | Whales | ISOWOA | The DNA storage codes filtered by ISOWOA increased by 2–18%, | Complexity is higher when computed with a large sequence. |
| Stephan et al. [86] | 2021 | Bee Colony, whale | HAW | Better accuracy, complexity, and computational time | Difficult to perform large datasets and its computation time is more |
3.10. Slime Mold Algorithm and its Application in Bioinformatics
In 2020, Li et al. [87] introduced the slime mold algorithm (SMA), a potent population-based optimizer inspired by the oscillation mode of slime mold in nature. The eukaryotic creature known as slime mold is distinct from other eukaryotes and has a unique life cycle that involves spore production and a free-living single-celled stage. Spores are commonly formed in fruiting structures with several cells or nuclei that are macroscopic in size and develop through fusion or aggregation. Slime molds were originally considered to be fungi, but that classification has since been eliminated. Although they don't fall into a single monophyletic group, they are categorized as Protista. The SMA has a distinctive mathematical model for many situations, especially real-world cases, and generates highly competitive results as well as quick convergence. The gradient-free SMA technique is used to model both positive and negative slime mold propagation wave feedbacks. The balance between global and local search drifts is stable, and the structure is dynamic. Below is a numerical representation of this slime mold behavior.
Where t is the number of current iterations.
 represents the optimal solution. During iteration,
represents the optimal solution. During iteration,
 and
and
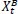 are two persons (slime mold) chosen at random from the population t.vb is a parameter with a value between -a and a.vc. ‘vc’ is a variable that decreases from 1 to 0. The letter W stands for the weight of slime mould. P is a parameter that can be determined using the following formula:
are two persons (slime mold) chosen at random from the population t.vb is a parameter with a value between -a and a.vc. ‘vc’ is a variable that decreases from 1 to 0. The letter W stands for the weight of slime mould. P is a parameter that can be determined using the following formula:
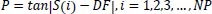
Where S(i) represents X. NP represents fitness and DF does have the best fitness.
The following is the formula for a:
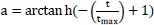
Where tmax denotes the number of iterations.
The derivation of the W formula is as follows:
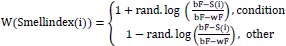
Smellindex = Sort(S)
The terms best fitness and worst fitness in the current population, respectively, stand in place of condition, which stands for those who are in the top half of fitness.
According to the following mathematical formula, the slime mold's location can be updated:
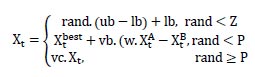
Where ub and lb represent the search space's upper and lower limits, respectively.
3.10.1. Slime Mold Algorithm (SMA)
Step 1: First slime molds positions are initialized.
Step 2: Calculate the fitness of all slime molds W by Eq. (17)
Step 3: Calculate each search position (Xi).
Step 4: Update parameters P, ub and vc.
Step 5: Update positions of slime mould by Eq. (18).
Step 6: Repeat steps 4 and 5 until the termination requirements have been met.
Step 7: Finally return the best Fitness and Xt
3.10.2. Literature Survey on SMA and its Application in Bioinformatics
This section examines the current innovative SMA algorithms and their applications over computational biology research and development. Qiu et al. [88] introduced an enhanced SMA (GLSMA) algorithm based on Levy flight and Gaussian mutation. According to experimental results, the two tactics are critical in expanding SMA's global search and preventing it from sliding into the local optimum. Beginning with comparisons to DE, PSO, GWO, and other well-known algorithms, the value of the GLSMA approach is shown.
Second, when compared to other advanced swarm intelligence algorithms such as MPEDE, LSHADE, ALCPSO, and CLPSO, GLSMA finds the optimal solution faster. After that, BGLSMA is created by translating GLSMA into binary space using a transformation function and is then used to highlight selection problems in 14 frequently used UCI high-dimensional gene datasets to show how well GLSMA performs in real-world applications. It is clear that GLSMA, in the application of feature selection, still has good global search ability and can select fewer features with higher classification accuracy when compared to a great metaheuristic optimizer in terms of general average characteristics selected, average error rate, average fitness, and average cost. To solve this issue, Qiu et al. [89] introduced a wrapper gene selection technique based on the slime mold algorithm. SMA is a revolutionary approach to feature selection with numerous applications. Their study expands on the original SMA by merging the Cauchy mutation mechanism with the crossover mutation technique based on differential evolution (DE). The transfer function then solves the gene selection problem by converting the binary continuous optimizer. ISMA, the continuous form of the approach, is initially used to study 33 classical continuous optimization tasks. The effect of the discrete version, or BISMA, was then explored further by comparing it to other gene selection methodologies using 14 gene expression datasets. The experimental results reveal that the continuous version of the algorithm achieves the optimal balance of local and global search capabilities, whereas the discrete version of the algorithm gets the greatest accuracy when selecting the fewest genes. Ewees et al. [90] proposed a modified slime mold algorithm based on the Marine Predators Algorithm (MPA) operators as a local search technique. This strategy enhances the SMAMPA technique's convergence rate while minimizing the attraction to local optima. The effectiveness of SMAMPA is tested using 20 datasets, and the findings are compared to cutting-edge FS approaches. It is also feasible to assess how adaptable SMAMPA is to real-world situations by employing it as a quantitative structure-activity relationship (QSAR) model. The results demonstrate how the developed SMAMPA method outperforms other approaches in terms of lowering the size of the datasets under test. Table 10 shows the summary of the literature survey on DNA sequencing by using SMA application on bioinformatics with advantages and disadvantages.
According to Fig. (2), the number of papers published in Scopus exceeds that of other publications. Furthermore, Figs. (2a and 2b) show the distribution of presented articles relevant to the bio-inspired computing technique from 2003 to the present year 2022. According to the findings, the number of published papers has been steadily increasing from 2003 to the present. However, the number of articles published in 2022 is noticeably higher than in previous years. According to the findings, the use of bioinspired computing techniques in various research and studies is expanding significantly year after year.
4. OPEN RESEARCH ISSUES IN BIOINFORMATICS
Gene expression is a biological process that converts DNA instructions into functional molecules called proteins. Not all cells in a living organism require proteins. Complex molecular mechanisms are required to switch the genes on and off. If it does not occur, diseases and illnesses will develop. To evaluate gene expression to diagnose, classify, and predict a disease or condition, biomedical research usually uses the DNA microarray approach. To predict how a drug or therapy will respond, DNA microarray data is also employed. DNA microarrays include cDNA (complementary Deoxyribose nucleic acid), SNP (single nucleotide polymorphism), and CNV (Copy Number Variation) microarrays [50]. Making cDNA, or DNA intones, necessitates the use of single-stranded RNA. A single nucleotide polymorphism (SNP) is a change in a DNA sequence that only happens once [91]. CNV is a phenomenon in which different portions of a genome are repeated from person to person. For assessing gene expression, a range of current tools are available. The Affymetrix Gene Chip and the cDNA bicolor glass slide are the most often used tools. To obtain the essential knowledge of gene expression with great precision, many hurdles and limits must be overcome. The following are the major challenges:
| Authors | Year of Publication | Inspiration | Methodology | Advantage | Disadvantage |
|---|---|---|---|---|---|
| Qiu et al. [88] | 2022 | Slime Mold | GLSMA | Selected features with higher classification accuracy | Evaluation time of quality of features is a complex process |
| Qiu et at [89]. | 2022 | Slime Mold | SMA | Highest accuracy when selecting the least number of genes | It does not achieve better overlap scores |
| Ewees et al. [90] | 2022 | Slime Mold | SMAMPA | To reduce the dimension of the tested datasets | Difficult to perform large datasets and its computation time is more |
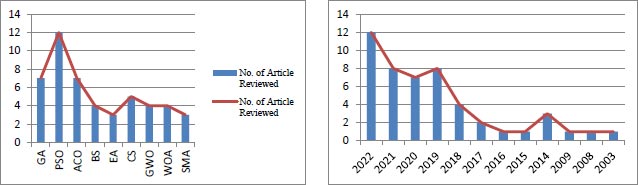
4.1. Curse of Dimensionality
The overfitting of a learning model is an important research topic in machine learning [50], and goes into great depth about the plague of dimensionality. Microarray data is typically high-dimensional, with hundreds to thousands or more properties. Microarray data handling is a difficult task. New storage methods are necessary to store such enormous volumes of data [54].
4.2. The Gap between Biologists and Researchers
Genetic research still has many research gaps due to the wide gap between scientists, biologists, and medical professionals. Because of the aforementioned gap, the chances of discovering the optimal tactics and approaches are relatively less.
4.3. Duplicate and Mislabeled Data
Due to the inconsistent scanning of data, imbalance and mislabeled data are the most common issues in microarray data. In most microarray datasets, there is a problem of class imbalance, in which one class dominates the entire dataset. It significantly reduces a learning model's generalization ability when it is trained on data that is unbalanced and incorrectly classified. When determining the effectiveness of the feature set, duplicated and redundant data are a significant concern [39, 50], much like the aforementioned issues.
4.4. Recovery of the Biological Information
The primary goals of genomic research are to gather biological data and identify changes in gene expression. There are several clinical difficulties, both clinically and physiologically. The challenge is that not everyone will have cutting-edge technology to record important occurrences. Changes in expression are also extremely modest and difficult to detect using analytical methods in other biological processes. The results might have been contaminated and incorrect due to the different strategies used in the experimental setup, data access, investigation, and chemical formulation.
4.5. The Research Prospects of this Study in this Field
4.5.1. Improved Diagnostic Models for Rare Genetic Disorders
Genetic diseases can be classified as either monogenic or polygenic. Monogenic diseases are extremely rare and are caused by mutations in a single gene. They are handed down from generation to generation. In contrast, polygenic disorders are more common and are brought on by mutations in many genes. Such inherited diseases have recently become excessively common. Accurate disease diagnosis will benefit from the application of bio-inspired approaches in ML classification and prediction models.
4.5.2. Cancer Diagnosis and Prediction
As cancer is a complicated illness with several subtypes, it is essential to identify patients as soon as possible to provide them with the best therapeutic care. Many studies have been done on bioinspired approaches used in bioinformatics and machine-learning applications since it is crucial to discriminate between high and low-risk patients. To develop classification and prediction models, machine learning models such as support vector machines (SVM), artificial neural networks (ANN), and Bayesian networks (BN) are essential.
4.5.3. Comparison of Multiple Sequences Alignment
When applied to gene expression data, individual machine-learning models will produce better outcomes. The effectiveness of hybrid techniques, on the other hand, has been demonstrated in many applications. More research is required on the topic of combining various clinical reports and hybrid approaches. Although challenging and time-consuming, it will result in superior outcomes.
4.5.4. Gene sequence data analysis for pharmacogenomics
It is essential to predict how medicine will respond to a genetic anomaly or disease. Outstanding efforts have been made recently to detect cancer sensitivity and response. However, the main obstacle to developing a drug analysis technique is the high dimension and small sampling size. Machine learning with feature selection techniques aids in minimizing dimensions and improving the accuracy of predicting drug reactions.
5. SUMMARY AND DISCUSSION
According to the study of the literature included in this survey, bio-inspired computing approaches are employed to solve challenging optimization problems in practically all domains of bioinformatics. In academic and business contexts, the meta-heuristic swarm intelligence optimization technique is becoming increasingly popular. The results of this literature survey indicate that, in addition to the domains of bioinformatics covered in this survey, the use of bioinspired computing approaches is surely expected to increase in a variety of other relevant fields (life science, electrical and power systems, computer engineering, aeronautical engineering, and construction engineering). Researchers from around the world have recently developed several algorithms based on bio-inspired methods to address a variety of engineering optimization issues, including the binary-based technique, fuzzy and neuro fuzzy-based methods, Levy flight-based methods, chaotic and antagonistic based learning methods, and more. The reviews based on bioinspired computing techniques include some of the most prevalent methodologies for resolving contemporary optimization problems. Increasing bioinspired methods are being used in numerous bioinformatics fields today. Bioinspired techniques are employed by 57% in computer science, 25% in electrical engineering, 13% in applied mathematics, 4% in aeronautical engineering, and 1% in construction engineering, as shown in Table 11. As a result, nature-inspired approaches are becoming more popular, followed by computer science and bioinformatics. The parametric comparisons across the works in this study suggest that most classic benchmark approaches, such as GA, EA, PSO, ACO, CS, GWO, BS, WOA, and SMA, can be successfully used by employing bioinspired techniques. According to the classification of bioinspired computing techniques, this literature study found that 56% of studies in the literature are modification based, 30% are hybrid-based, and 14% are multiobjective based, as illustrated in (Fig. 3). The use of bioinspired approaches has produced good results across the board and demonstrated its superiority in the solving of complex optimization problems.
| Algorithms | Inspiration | Representation | Area of application |
|---|---|---|---|
| GA [31] | Evolution Theory | Permutation of elements, binary, real no, programme elements, data structures, trees, and matrix | Data mining and pattern reorganization, several criteria that are flexible optimization of websites, distributed detection selection limits in wireless sensor networks, path planning for robotic systems with computer assistance, the fixed mobility problem, multiple scheduling and task problems, the design of flight control systems, pattern recognition, training for the radial basis function, Software development issues, the need to reduce pollution levels in the industrial sector, the multi-objective vehicle routing problem, the mobile ad hoc network's minimal energy broadcast issues, problems with portfolio optimization, power system optimization, and the best e-learning learning path Web service selection, structure optimization, defect detection, molecular modeling, cutting stock problem, medication design, and the closest sting problem in bioinformatics, DNA sequencing. |
| PSO [39] | Coordinated movement of birds and fish school |
N-dimensional vector for the position, speed, and best state. | Iterated Prisoner's Dilemma and multimodal biomedical image registration, a multiclass database's ability to categorize instances, feature selection, the creation of online services, the design of educational curricula, and issues with power system optimization (economic dispatching), edge detection in noisy images, selecting the optimal machining parameters, scheduling issues, vehicle routing issues, tool life prediction in ANN, multi-objective, dynamic, constrained, and combinatorial optimization problems, QoS in ad hoc multicast, anomaly detection, colour image segmentation, sequential ordering problem, restricted portfolio optimization challenge, partial particle regeneration for data clustering, machine defect detection, unit commitment computation, and rule extraction from fuzzy neural networks Verification of the signature, DNA sequencing. |
| ACO [54, 55] | Ants foraging | Undirected graph | Quadratic Assignment Problem, TSP Problem (QAP) Scheduling problem in the manufacturing tasks. Data network routing, shortest path problem in a dynamic network, parallel processing implementations, and ongoing optimization. Problems of protein folding, data mining, classification, digital image processing, agent-based dynamic scheduling, graph coloring, set covering, and DNA sequencing |
| BS [63] | Echolocation Behavior of microbats |
n-dimensional vector for the position, speed, best state, graph | Optimization, modeling, control, workshop scheduling problem, vehicle path, drug waste collection and distribution, data mining classification problems, digital image processing, graph coloring, set covering, and protein folding, DNA sequencing, global search, and local search. |
| EA [67] | Evolution Theory | Real-valued vectors | Image processing, computer face recognition, parameter estimation, task scheduling, structural improvement, gas turbine fault diagnosis evolution strategy, clustering problem with vehicle routing using a multi-parametric evolution method. |
| CS [71] | Cuckoo’s egg laying and breeding | Levey fight, an n-dimensional vector of position, speed, and the optimal state graph | Optimization, modeling, control, workshop scheduling problem, vehicle path, drug waste collection and distribution, classification problems in data mining, and digital image processing, agent-based dynamic scheduling, graph coloring and set covering, cloud computing, DNA sequencing, global search, and local search, vehicular Adhoc Network. |
| GWO [77] | Grey wolf and their behavior of the social hunting process | Coordinated movement of Grey wolf |
Scheduling problems, Manufacturing field, system modeling, frequency control problems, robotic path planning, image processing, optimization problem, clustering, feature selection, medicine and bioinformatics, and wireless sensor networks. |
| WOA [83] | Whale’s social hunting process | Coordinated movement Of Whale |
Dimensionality reduction and classifications problem, Feature Extraction, Feature selection, Clustering, Training of MLP network, Drug toxicity prediction, Male fertility rate categorization, DNA Sequence Optimization. |
| SMA [87] | Inspired by the oscillation mode of slime mold in nature | Graph | Data Analytics Problems, Global Optimization, Image segmentation, power flow controller, Feature selection, Solving TSP Problem, Multi-level thresholding, DNA Sequence Optimization. |
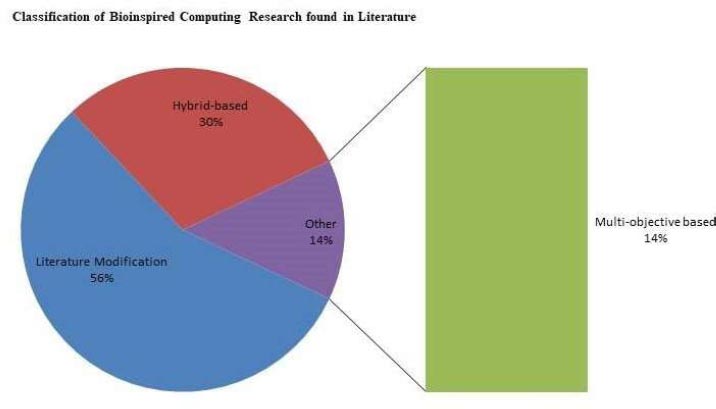
CONCLUSION
The bioinspired computing optimization algorithms are more application-specific, requiring different parameter values for different applications. It enables a wide range of applications to obtain specific global optimal solutions. Machine learning (ML) has offered to play a vital role in combining bioinformatics and various big data components, which are the most recent applications of ML and bioinspired computing approaches that have been highlighted in the literature. These new analytical techniques have helped in identifying both the symptoms and signs of various illnesses as well as in predicting epidemics. Additionally, contact tracing, genetic research, and the development of medicines for a wide range of diseases have all been made possible by machine learning-based bioinspired big data analytics, which has displayed excellent performance. In bioinformatics, a variety of bioinspired optimization algorithms are applied. This review article, “A Study of Bioinspired Computing in Bioinformatics: A State-of-the-Art literature Survey” includes a variety of bioinspired optimization algorithms in bioinformatics based on genetic algorithm (GA), particle swarm optimization algorithm (PSO), ant colony optimization algorithm (ACO), evolutionary algorithm (EA), cuckoo search algorithm (CS), and grey wolf optimization algorithm (GWO), Whale Optimization Algorithm (WOA), Slime Mold Algorithm (SMA), which can solve the data set optimization of DNA sequencing and disease diagnosis and prediction. These algorithms can be used to optimize data sets because of their capacity to solve real-world challenges. These bioinspired algorithms may outperform conventional bioinformatics algorithms in such a scenario. These strategies may be applied to optimize data sets used in bioinformatics since they are successful at enhancing hardware design parameter optimization. Another issue identified in the study is determining how to accurately express the sequence quality and evaluate DNA sequence classification. The two most important aspects of DNA sequence clustering are determining how to identify distinct subsequences from a DNA sequence and mining patterns, which generate a flood of possible sequence patterns and require a formidable amount of time and space.
In the future, it might be expanded to data set optimization in software applications and analysis of larger DNA subsets, proposing better complexity measures adapted for the larger sequence of DNA and offering insight on pattern analysis for various types of biological and medical data, where the set parameters correspond to the parameters chosen in each of these algorithms.
AUTHORS CONTRIBUTIONS
Akshaya Kumar Mandal (https://orcid.org/0000-0003 -0955-4096): Data correction, Resources, Writing – review & editing – original draft. Pankaj Kumar Deva Sarma (https://orcid.org/0000-0002-9748-3787): Conceptualization, Supervision, review & editing. Satchidananda Dehuri (https://orcid.org/0000-0003-1435-4531): Conceptualization, Supervision, review & editing.
LIST OF ABBREVIATIONS
| GA | = Genetic Algorithm |
| PSO | = Particle Swarm Optimization Algorithm |
| ACO | = Ant Colony Optimization Algorithm |
| EA | = Evolutionary Algorithm |
| CS | = Cuckoo Search Algorithm |
| GWO | = Grey Wolf Optimization Algorithm |
| WOA | = Whale Optimization Algorithm |
| SMA | = Slime Mold Algorithm |
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by Dr. Satchidananda Dehuri, Professor of Computer Science (Erstwhile P. G. Department of Information and Communication Technology), Fakir Mohan University would like to thank SERB, Govt. of India for financial support under Teachers’ Associateship for Research Excellence (TARE) fellowship vide file no: TAR/2021/000065 for the period 2021–2024.
CONFLICT OF INTEREST
The authors declare no conflict of interest financial or otherwise.
ACKNOWLEDGEMENTS
We gratefully acknowledge the anonymous referees for specific suggestions that aided in improving the presentation of this article.

