
EZYDeep: A Deep Learning Tool for Enzyme Function Prediction based on Sequence Information
Authors Info & Affiliations
Abstract
Introduction:
Enzymes play a crucial role in numerous chemical processes that are essential for life. Accurate prediction and classification of enzymes are crucial for bioindustrial and biomedical applications.
Methods:
In this study, we present EZYDeep, a deep learning tool based on convolutional neural networks, for classifying enzymes based on their sequence information. The tool was evaluated against two existing methods, HECNet and DEEPre, on the HECNet July 2019 dataset, and showed exceptional performance with accuracy rates over 95% at all four levels of prediction.
Results:
Additionally, our tool was compared to state-of-the-art enzyme function prediction tools and demonstrated superior performance at all levels of prediction. We also developed a user-friendly web application for the tool, making it easily accessible to researchers and practitioners.
Conclusion:
Our work demonstrates the potential of using machine learning techniques for accurate and efficient enzyme classification, highlighting the significance of sequence information in predicting enzyme function.
1. INTRODUCTION
Enzymes are proteins that have a specific biological activity: they catalyze biological reactions. Catalysts are tools that can substantially speed up these processes. At the conclusion of the reaction, they are regenerated and act at low concentrations. The vital functions of organisms are crucially maintained by enzymes. They control numerous other chemical processes that are strongly related to the process of life, including metabolism, nutrition, and energy conversion. Enzymes are utilized in a variety of processes, including industrial synthesis, human health, and environmental remediation [1, 2].
Precision in the selection of enzymes for bioindustrial or biomedical applications is therefore crucial and has become a focus of great medical, environmental, and industrial importance. Most enzymes are proteins, while others are ribonucleic acids. Until now, research on the prediction of classes and subclasses of enzymes has always been focused on the enzyme whose chemical nature is the protein; in other words, when computer models are used to classify enzymes, the method of feature extraction adopted is always protein-specific. To facilitate the in-depth study of enzymes, it has become important to classify and name enzymes internationally. “International Union of Pure and Applied Chemistry” (IUPAC) and “International Union of Biochemistry and Molecular Biology” (IUBMB) have created the “IUPAC-IUBMB Joint Commission on Biochemical Nomenclature” (JCBN) and the “Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB)”. It is the NC-IUBMB that maintains the nomenclature and classification of enzymes [3].
The latter created an international commission called the Enzyme Commission, which is tasked with developing a nomenclature for enzymes. The Commission has divided the enzymes into 7 major classes depending on their reactions (see Table 1 to look at the classes) [4]. The EC nomenclature is made up of four parts that identify respectively the main class, subclass, sub-subclass, and substrate class of the enzyme [5]. It is worth mentioning that in recent years, many classifiers capable of classifying enzymes have appeared. Most of the classifiers designed by researchers can classify enzymes at the subclass level [6]. As enzymes were divided into 6 major classes according to the type of reaction catalyzed, a seventh class, the translocase, was added in 2018 [4]. Most prediction methods divide the enzymes into just six classes [7-11]. Functional laboratory identification approaches have been used to discover the function and class of enzymes, although this form of experiment is costly and time-consuming [12, 13]. Consequently, it is appropriate to classify enzymes using bioinformatics technologies and deep learning methods.
| EC Number First Digit | Name |
|---|---|
| 1 | Oxidoreductases |
| 2 | Transferases |
| 3 | Hydrolases |
| 4 | Lyases |
| 5 | Isomerases |
| 6 | Ligases |
| 7 | Translocases |
With the development of bioinformatics and deep learning [14, 15], researchers have designed many models for enzyme class prediction [16]. In 2003, F. Y. Hunt et al [17] introduced an algorithmic approach to facilitate sequence alignments between a query sequence and a database of sequences. The primary aim of this approach was to identify highly similar proteins that are already annotated, under the assumption that information about the biological function or structure of a protein can be gained through such identification. In 2009, Nasibov et al [18] adopted the K-nearest neighbor (KNN) classification method, which is a machine-learning classification algorithm used for classification and regression tasks. It works by finding the K closest data points in the training set to a given test point, and the class of the test point is determined by a majority vote of the K-nearest neighbors. In 2010, Qiu et al [19] used the support vector machine (SVM), achieving good results. In 2018, Yu Li et al [20] used a combination of Convolutional neural network (CNN) and long short-term memory (LSTM) to build their classification model. In addition, in order to achieve better prediction results, researchers usually combine various feature extraction and classification methods in their prediction process. For example, Shen et al [21] combined functional domain (FunD) and scoring matrix (PsePSSM) to extract features in 2009. Wang et al [22] combined composition, transition, and distribution (CTD) and pseudo amino acid composition (PseAAC) to extract features and ranked sequences with the combination of (RAkEL-RF) and multi-label KNN (MLKNN) methods in 2014. In 2018, Yu Li et al [20] combined one hot encoded protein sequence, position-specific scoring matrix (PSSM), solvent accessibility information, secondary structure information predicted by DeepCNF [23], and functional domains. In 2020, Safyan Aman Memon et al [24] combined the protein sequence, PSSM, disordered regions, protein secondary structure, solvent accessibility, amino acid composition, and functional domains.
Our contribution consists in proposing a new approach in which artificial intelligence is the pillar. It consists in using natural language processing (NLP) [25] with deep learning to determine the function of enzymes based on their primary structure. we chose the protein sequence as the only training data in order to avoid involving erroneous data from a false prediction (for example predicted protein secondary structure...) so that the latter is the only data necessary to predict the function. The organization of the rest of the paper is as follows: The research methodology is described in Section 2. Section 3 presents the results and discussion. The last section concludes this paper and discusses future work.
2. MATERIALS AND METHODS
2.1. Datasets
The dataset was obtained from the UniProt Knowledge Base, the protein sequences collected are publicly available at (https://www.uniprot.org/). This database has two types of sequences: reviewed and unreviewed sequences. We chose only the reviewed ones, but before proceeding to download, we selected only three columns, which are Sequence, EC number, and Sequence Length. These three columns represent the data, the label, and the length column have been used for data cleaning purpose. The downloaded file format was tab-separated values (TSV). The file contained initially 568,002 sequences (October 10, 2022). To note, sequences with no EC number are non-enzyme proteins.
2.2. Used Environment
EZYDeep tool is developed using Python as a programming language. Many libraries are involved in the programming process: we start with Pandas that we used in order to clean our datasets. The 2nd main library was Tensorflow that we used to build our model, then sklearn which we used to measure the efficiency of the model. Finally, we used Django framework to build the website where the tool is available at (http://rs.umc.edu.dz:8000/).
Additionally, EZYDeep models are trained on Kaggle, as it offers a powerful system equipped with GPUs (2x Nvidia Tesla T4 with 16 GB of VRAM).
2.3. Data Cleaning
The dataset went through many steps of data cleaning, starting by deleting all sequences with a length higher than 1,500 aa or lower than 50 aa, followed by deleting sequences with unknown amino acids (represented by X character) and uncommon amino acids (O, J, U, Z, B). Then, all the sequences with incomplete EC numbers that include a dash (–) and the ones with multiple EC numbers were deleted. The number of sequences after this cleaning decreased to 439,301 sequences. After the cleaning phase, we made a copy of our dataset so it can be dedicated to enzyme models only.
In the copy containing all the 439,301 sequences, we added a column that contains a binary labeling: enzyme or non-enzyme for every sequence, as non-enzyme sequences have no EC number. Level 0 of the classification will be based on that column, as it will classify the sequences into enzymes or non-enzymes. Table 2 shows the detail of the Level 0 dataset.
| Classes | Number of Sequences Per Class |
|---|---|
| Enzyme | 204,008 |
| Non-enzyme | 235,293 |
In the 2nd copy, all the non-enzyme sequences have been deleted. The number of the remaining sequences is 204,008.
Three columns have been added to this dataset, all based on the EC number column:
The 1st column contains the 1st EC number represented by values from 1 to 7 as there are 7 enzyme classes. Level 1 of the classification will be based on that column, Table 3 shows the details about the number of sequences by classes at the 1st level of classification.
| Classes | Number of Sequences Per Class |
|---|---|
| 1 | 23,331 |
| 2 | 72,684 |
| 3 | 41,246 |
| 4 | 18,946 |
| 5 | 12,178 |
| 6 | 24,200 |
| 7 | 11,423 |
The 2nd column contains the 1st two EC numbers which means the enzyme class and subclass. Level 2 of the classification will be based on that column.
The 3rd column contains the 1st three EC numbers which mean the enzyme class, its subclass, and its sub-subclass. Level 3 of the classification will be based on that column.
The number of classes in level 2 and level 3 are 64 and 193 respectively.
2.4. Data Encoding
As deep learning models cannot handle directly alphabetic characters (raw text), the sequences and labels we have must be encoded into a numeric format. In order to do so, we started by encoding the labels using a one-hot encoder that consists in encoding a label with n states on n bits of which only one takes the value 1, the number of the bit being 1 being the number of the state taken by the label. This will convert our classes into arrays built with 0 and 1 (Table 4).
| - | Enzyme | Non-enzyme |
|---|---|---|
| Enzyme | 1 | 0 |
| Non-enzyme | 0 | 1 |
The following step was to encode the sequences into a numeric format. The used method is Character-level Tokenization. This method consists in splitting our sequences into individual characters then every character gets its own numeric value. The tokenization will convert our sequences into arrays, which, unlike label conversion, will be built with numbers from 1 to 20 (as the number of amino acids is 20). Another step is necessary, which is sequence padding. This method adds zeros to every array whose size is inferior to 1500 so at the end all the arrays will be of the same size.
For every level of classification, the used dataset has been divided into 2 sets in a 9:1 ratio. The first part was used so the deep learning model can learn from it, the second part was used to test the efficiency of the model.
2.5. Proposed Model
Four models have been built, one model for every level of classification (from level 0 to level 3).
As tokenization gives random values to sequence characters, which means similar amino acids can have very distinct values, a character embedding must take place. So, the first layer of our model is the embedding layer, whose role is to convert every integer representing an amino acid into a vector, the result of this is having two dimension arrays representing every protein/enzyme sequence. Those array values are learned along with the model itself. This process is very efficient when dealing with huge datasets like ours, as it allows the values of vectors representing similar amino acids to be reconciled which captures relationships that are very difficult to capture.
The second layer is the convolutional layer, which receives the output of the embedding layer. This layer was built with a number of filters varying from 64 to 512, and its kernel size varied from 3 to 32. Despite the usual use of small kernel size, using bigger ones led to much better-performing models as some papers mentioned [26].
The third layer is a max polling layer whose goal is to reduce the size of learned features, consolidating them only to the most essential elements.
The fourth layer is the flattening layer, whose purpose is to convert the two dimensions vector outputted by the max pooling layer into a one-dimension vector which is fed into several fully connected layers with dropout regularization so that the model can easily define the relationship between the values of the data and their labels.
Both binary cross entropy and categorical cross entropy have been used. The first is for the binary classification (enzyme and non-enzyme), the last is for the multiple classification (classification based on EC number from level 1 to level 3).
2.5.1. Model Learning and Evaluation
Once the model is built, we instantiate it and start the training. When the training is over, we proceed to the evaluation of the model, i.e., evaluating its accuracy.
2.5.2. Hyper-parameters Tuning
After evaluating the model, we tried many combinations of hyper-parameters in order to find the model with the best prediction accuracy. Some of those hyper-parameters are the embedding dimension, the number of convolutional layer filters, the kernel size of the convolutional layer, the pool size of the max pooling layer, as well as the number of nodes of the dense layers. The used parameters are displayed in Table 5.
| Hyper-parameter | Tested Values |
|---|---|
| Embedding dimension | 32, 64, 128, 256 |
| Convolutional layer filters | 64, 128, 256, 512 |
| Kernel size | 3, 4, 6, 8, 16, 32 |
| Pool size | 3, 4, 6, 8, 16, 32 |
| Neurons | 64, 128, 256, 512, 1024, 2048 |
| Batch size | 64, 128, 256 |
| Dropout | 0.025, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6 |
| Epochs | 30, 50, 70, 100 |
2.6. Web Application
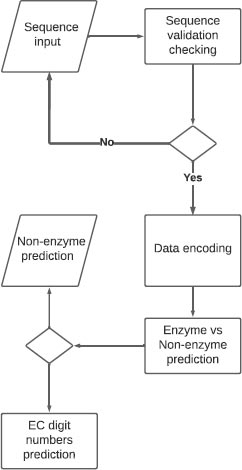
The website built is basically one web page where the user can input a protein sequence. A backend function checks if the input represents a real protein sequence by checking many criteria: sequence size, the first amino acid of the sequence, and the amino acids composing the sequence. The inputted sequence is encoded the same way our dataset sequences were encoded. The encoding output is then passed to the models so the prediction can be made. The results are then displayed to the user in a table (Fig. 1).
2.6.1. Model Overview
EZYDeep, the tool presented in this paper, is a deep learning method based on convolutional neural networks. The experiments were performed on 4 levels which are: level 0: Enzyme and non-enzyme, level 1: first digit EC number prediction, level 2: second digit EC number prediction, level 3: third digit level EC number prediction. We performed some tests on level 4 too which is the fourth and last digit of the EC number but as the majority of 4th-level classes have a number of sequences inferior to 5, the experiment did not yield satisfactory results.
2.6.2. Evaluation Measure
To assess the quality of the predictions, we employed several commonly adopted metrics [27, 28]. These metrics provide a comprehensive evaluation of the performance of our enzyme classification models. The evaluation metrics used are as follows:
1- Accuracy: This metric calculates the ratio of correctly classified instances (True Positives and True Negatives) to the total number of instances evaluated (True Positives, True Negatives, False Positives, and False Negatives). It is defined as:
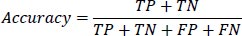 |
2- Recall: Also known as the true positive rate, recall measures the proportion of true positive instances correctly identified by the model. It is defined as:
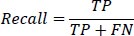 |
3- Precision: Precision measures the proportion of true positive instances out of all instances predicted as positive. It is defined as:
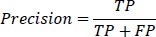 |
4- F1-score: The F1-score is the harmonic mean of precision and recall, providing a balanced measure of the model's performance. It is defined as:
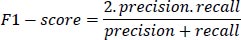 |
Where:
True Positive (TP): means the actual value and predicted value are the same.
True Negative (TN): means the sum of values of all columns and rows except the values of that class that we are calculating the values for.
False Positive (FP): means the sum of values of the corresponding column except for the TP value.
False Negative (FN): means the sum of values of corresponding rows except for the TP value.
In our experimentation, we conducted a two-step approach. First, we performed a simple test by randomly splitting the dataset into 90% for training and 10% for testing. This initial test provided a baseline assessment of the model's performance.
Subsequently, we employed cross-validation with 10 folds on the entire dataset. This process involved dividing the data into ten equal parts, iteratively training the model on nine parts, and testing the remaining part. By repeating this process ten times, we ensured that each part of the data was used for both training and testing. This cross-validation approach enabled us to obtain a more robust evaluation of the model's performance.
By employing this experimental setup, we obtained comprehensive insights into the effectiveness and generalizability of our enzyme classification model.
3. RESULTS AND DISCUSSION
As mentioned before, the models are tested on 10% of the dataset. The test method is based on the confusion matrix.
Table 6 shows the performance of the EZYDeep models on four different levels of classification, with evaluation metrics including accuracy, precision, recall, and F1-score. At Level 0, the model achieved an accuracy of 97.04% with high precision, recall, and F1-score. For Level 1 classification, the model achieved an accuracy of 97.35% with high precision, but lower recall and F1-score compared to Level 0. For Level 2 classification, the model achieved a lower accuracy of 96.41% with decreased precision, recall, and F1-score. At the highest level of classification (Level 3), the model achieved an accuracy of 95.83% with a high precision, but a significant decrease in recall, resulting in a lower F1 score compared to the other levels. These results suggest that the model performs well for broader classifications.
| - | EZYDeep | |||
|---|---|---|---|---|
| - | Acc | Precision | Recall | F1 |
| Level 0 | 97.04 | 97.39 | 97.44 | 97.35 |
| Level 1 | 97.35 | 98.25 | 97.13 | 97.08 |
| Level 2 | 96.41 | 94.46 | 93.53 | 93.95 |
| Level 3 | 95.83 | 95.36 | 89.10 | 91.63 |
In addition to the test results obtained from the 10% dataset, we further conducted 10-fold cross-validation to assess the performance of our EZYDeep models. The evaluation metrics used were accuracy, precision, recall, and F1-score. Table 7 presents the performance of the models at four different levels of classification.
| - | EZYDeep | |||
|---|---|---|---|---|
| - | Acc | Precision | Recall | F1 |
| Level 0 | 97.01 | 97.04 | 97.01 | 96.90 |
| Level 1 | 97.06 | 97.08 | 96.06 | 97.06 |
| Level 2 | 96.13 | 96.22 | 96.13 | 96.14 |
| Level 3 | 95.46 | 95.79 | 95.46 | 95.53 |
The performance measures in Table 6 demonstrate a strong correlation between the evaluation metrics. At Level 0 classification, the model achieved an accuracy of 97.01%, with a precision of 97.04%, recall of 97.01%, and an F1-score of 96.90%. Similarly, at Level 1 classification, the model achieved an accuracy of 97.06%, with a precision of 97.08%, recall of 96.06%, and an F1-score of 97.06%. Moving to Level 2 classification, the model achieved an accuracy of 96.13%, with a precision of 96.22%, a recall of 96.13%, and an F1-score of 96.14%. Finally, at the highest level of classification (Level 3), the model achieved an accuracy of 95.46%, with a precision of 95.79%, recall of 95.46%, and an F1-score of 95.53%.
These results suggest a consistent trend, indicating that as the classification becomes more specific (higher levels), the model encounters challenges in accurately identifying instances belonging to more specific enzyme classes. This leads to a decrease in precision, recall, and subsequently affects the F1-score, while the accuracy remains relatively high.
The inclusion of cross-validation allows for a more comprehensive evaluation of the model's performance, taking into account variations in the dataset splits and providing a more robust assessment of its generalization capabilities. The strong correlation observed between the evaluation metrics further validates the model's performance across different levels of classification.
The consistent trend observed in the performance measures indicates the model's ability to achieve high accuracy at broader classification levels. However, as the classification becomes more specific, the model encounters challenges in accurately identifying instances belonging to more specific enzyme classes. To gain further insights into the model's limitations, we analyzed the misclassified sequences, which shed light on potential factors contributing to these errors.
When analyzing the misclassified sequences, several factors could have contributed to the incorrect classification. One possible reason is the presence of ambiguous or overlapping features within the dataset. Enzymes with similar characteristics or functional properties may share common sequence patterns, making it challenging to distinguish between them accurately. Additionally, the model's performance may be influenced by the quality and representativeness of the training data. As certain enzyme classes are underrepresented or inadequately represented in the training set, the model may struggle to learn their distinguishing features effectively. Furthermore, the complexity of the classification task and the inherent variability within enzyme families can also pose challenges. Some enzymes may exhibit diverse functional properties within the same class, leading to misclassification due to subtle differences in their sequences.
Addressing these challenges could involve refining the feature representation used by the model, augmenting the training data with more diverse and representative samples, and exploring more advanced machine-learning techniques or ensemble approaches. By investigating and understanding the reasons for misclassifications, we can enhance the accuracy and robustness of the classification model in future iterations.
Overall, the results obtained from the evaluation metrics and the analysis of misclassified sequences provide valuable insights into the performance and limitations of our EZYDeep models. These findings can guide further research efforts toward developing more accurate and reliable enzyme classification models, ultimately contributing to a deeper understanding of enzyme functionality and facilitating various biotechnological applications.
4. COMPARISON WITH PREVIOUS METHODS
In our study, we aimed to develop a tool for predicting non-enzymes and enzyme functions based solely on their sequence information. To validate its performance, we conducted a comparison with two existing tools, HECNet [24] published in 2020, and DEEPre [20,28] published in 2018, which incorporate additional information, such as protein secondary structure, in addition to non-enzymes and enzymes sequences for prediction.
To evaluate the performance of these tools, we used the HECNet July 2019 dataset, consisting of 12,889 enzymes and 30,374 non-enzymes instances. To compare the performance of the tools, we used the accuracy and F1 score as evaluation measures. The results, shown in Table 8, demonstrate that our tool outperformed both HECNet and DEEPre, achieving higher accuracy and F1 score on the July 2019 dataset.
| - | EZYDeep | HECNet | DEEPre | |||
|---|---|---|---|---|---|---|
| - | Acc | F1 | Acc | F1 | Acc | F1 |
| Level 0 | 98.56 | 98.25 | 94.0 | 94.1 | 95.9 | 95.9 |
| Level 1 | 98.76 | 98.80 | 93.6 | 82.8 | 91.8 | 87.3 |
| Level 2 | 98.23 | 94.59 | 93.5 | 70.6 | 88.8 | 63.4 |
| Level 3 | 98.11 | 97.04 | 93.3 | 74.1 | 86.9 | 53.3 |
This comparison (Table 8) highlights the strengths of our tool, which is able to effectively classify and predict non-enzymes and enzyme functions based solely on their sequence information. However, it also highlights some of the limitations of our tool in comparison to previous methods that incorporate additional information. In the future, it may be worth exploring the integration of complementary data sources, to further improve the accuracy of the prediction.
Overall, our results demonstrate the potential of using deep learning for non-enzymes and enzyme functions prediction based on sequence information, and suggest promising avenues for future research in this area.
Moreover, our results suggest that deep learning approaches could be used to predict enzyme functions even for sequences that have not yet been experimentally characterized, paving the way for more efficient enzyme annotation and drug discovery.
CONCLUSION
In conclusion, this study presents a deep learning tool for classifying enzymes based solely on sequence information. The tool, designed using a sequential model with convolutional and dense layers in TensorFlow, showed exceptional performance when evaluated against two existing methods, HECNet and DEEPre, on the HECNet July 2019 dataset. The results revealed that our tool outperformed both HECNet and DEEPre in terms of accuracy and F1 score, underscoring the potential of using sequence information alone to accurately classify enzymes. Notably, our tool achieved a very high accuracy rate of over 95%, indicating its effectiveness in predicting enzyme function.
Our comparison with state-of-the-art enzyme function prediction tools proved that our tool performs significantly better than other methods at the four levels of prediction. This indicates that our tool has the potential to become a valuable resource for predicting enzyme functions, potentially speeding up the process of enzyme annotation.
This work demonstrates the capability of deep learning to capture functional information contained within enzyme sequences and highlights the significance of sequence information in predicting enzyme function. While our tool showed promising results, there is still room for improvement, and future studies may consider incorporating additional information to enhance the accuracy of enzyme classification.
In summary, this study has significant implications for the field of enzyme classification and for the advancement of biomedical research. The success of our tool and its availability through a web application highlights the potential of using machine learning techniques to aid in the prediction and understanding of enzyme function and provides a foundation for future research in this area.
LIST OF ABBREVIATIONS
| NLP | = Natural Language Processing |
| FunD | = Functional Domain |
| IUBMB | = International Union of Biochemistry and Molecular Biology |
| IUPAC | = International Union of Pure and Applied Chemistry |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No humans and animals were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data and supportive information are available within the article.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

