
Structure and Ligand-based In Silico Studies towards the Natural Inhibitors against Receptor Recognition Spike Protein of SARS-CoV-2
Authors Info & Affiliations
Abstract
Aim
In silico exploration, identification, and therapeutic potential of natural bioactive compounds as antiviral agents are compared with established FDA-approved antiviral drugs. The main aim of this study is to harness the best of plant-based bioactive compounds in the ongoing battle against viral infections.
Objectives
To investigate the specific amino acid residues within the spike (S) protein of SARS-CoV-2 that exhibit active sites. Furthermore, our objective is to evaluate the efficacy of molecular interactions between the active site residues of spike (S) protein of SARS-2 with FDA-approved drugs and screened bioactive compounds as a potential therapeutic antiviral agent employing in silico approach.
Background
The ongoing global pandemic, attributed to the SARS-CoV-2 virus, commonly known as severe acute respiratory syndrome, continues to spread, giving rise to various emerging variants. These variants, identified as variants of concern or interest (VOC/I), revealed a significant threat to global health, contributing to the severity of the catastrophe.
Methods
We explored how natural bioactive compounds derived from plants and certain FDA-approved drugs interact with the spike (S) protein of the virus. To do this, we employed techniques called molecular docking and molecular dynamics simulation (MD & MD Simulation) to evaluate the antiviral potential of screened bioactive compounds against spike (S) protein.
Results
In this study, Withanolide B and A bioactive compounds revealed the best molecular interaction (binding affinity) with spike (S) protein the SARS-CoV-2, with binding energies of -8.6 and -8.3 kcal/mol, respectively.
Conclusion
Unlike usually prescribed drugs, Withanolide B & A, derived from Withania somnifera, commonly known as ashwagandha, were found to be potential inhibitors of the spike (S) protein. This study indicates and emphasizes the promising role of natural compounds in the combat against COVID-19.
1. INTRODUCTION
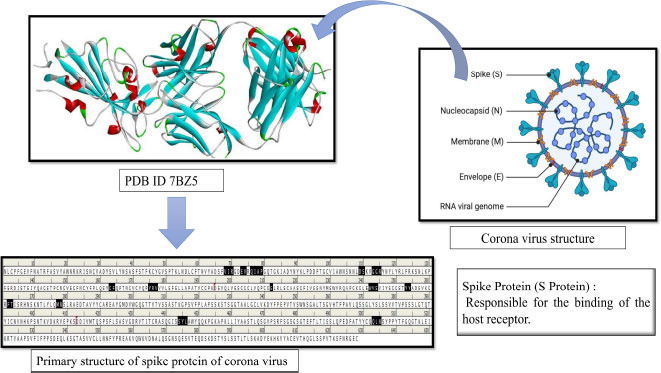
The global pandemic problem with SARS-CoV-2 and its variants is still a major health issue, causing huge mortality [1]. The spread of the virus has led to many questions about how to treat affected patients and keep them healthy. It all started in Wuhan, China, in December 2019, and since then, this dangerous virus, called SARS-CoV-2, has quickly spread around the globe, infecting people worldwide [2-6]. Taxonomically, the SARS-CoV-2 belongs to the Coronaviridae family and Nidovirales order and is a kind of virus with an Enveloped (E) protein and a single-stranded RNA (ssRNA). It is a tiny virus, about 40-60 nm in diameter, and it looks like it has a crown on its surface, hence the name coronavirus [7, 8]. The coronavirus family has four main groups: alpha (α), beta (β), gamma (γ), and delta (Δ). The alpha (α) and beta (β) types are the ones that usually infect healthy persons, but we are not sure how the gamma (γ) and delta (Δ) modes of action yet. Interestingly, the newly emerged coronavirus shares 80% of its genetic material with the well-known SARS-CoV-2 virus, which first appeared in China in 2002–2003 [9, 10]. Recently, there have been different types of coronaviruses, like 229E (α-CoV), HKU1 (β-CoV), OC43 (β-CoV), and NL63 (α-CoV), causing mild infections in individual’s upper respiratory tracts [11, 12]. However, there are two types of the virus, SARS-CoV and MERS-CoV, that have raised serious health concerns, and the latest one is SARS-CoV-2 (Beta-coronavirus) [13]. SARS-CoV-2 is highly contagious and can be harmful to the lungs and other important organs like the heart, liver, kidneys, gastrointestinal tract, and central nervous system. It often leads to failure of multiple organs [14, 15]. Additionally, SARS-CoV-2 has four main protein parts: spike (S) protein, envelope (E), membrane (M), and nucleocapsid (N) [16, 17]. Various types of polyproteins, both structural and non-structural, are required for SARS-CoV-2 to multiply into more copies of itself (Fig. 1). Interestingly, the spike (S) protein attaches to a receptor protein called angiotensin-converting enzyme (ACE-2) on the cells of the host who got infected. The spike (S) protein cleaved into smaller parts, namely PPLA and IAB through viral proteases during pathogenicity established respectively. Additionally, the spike (S) protein is more important due to its mode of action, it acts as a virulence factor by interacting and adhering to ACE-2 proteins of host cells. Therefore, the spike protein of SARS-CoV-2 like a significant target could be hit by natural bioactive compounds to interfere with the pathogenesis mechanism of SARS-CoV-2 [18-20]. Currently, there are no FDA-approved drugs identified specifically for treating SARS-CoV-2 (COVID-19). In cases of SARS-CoV-2 infection, whether it is mild or severe, doctors usually focus on managing preliminary symptoms. They were commonly prescribed FDA-approved drugs like Remdesivir, EIDD-2801, Hydroxychloroquine, Dexamethasone, Rito navir, Favipiravir, and Chloroquine. However, these drugs have not shown effective results, and they might have side effects for patients [21]. Using an overdose of these aforementioned drugs in affected persons with corona virus has been associated with lower levels of anti-inflammatory substances, causing a problem known as cytokine storm syndrome (CSS) and acute respiratory distress syndrome (ARDS)., Dexamethasone, a type of corticosteroid, is one drug that has been used to reduce mortality in the early stages of treatment.
While the adverse effects of dexamethasone are most frequently associated with anxiety, weight gain, fluid retention, hormonal imbalance, and insomnia [22]. The commonly used drugs such as hydroxychloroquine and chloroquine prevent virus and host cell interaction process and lead to control the activation of T-cells [23-25]. Drugs like Remdesivir, Favipiravir, and EIDD-2801 interfere with the activity of the RNA-dependent RNA polymerase of SARS-CoV-2, while the drug Ritonavir inhibits the activity of viral proteases, specifically cysteine protease PLpro and Mpro. Interestingly, natural herbs and their derivatives, known for minimal side effects and biocompatible, have also shown promising potential bioagents against viral infections [26-28]. Certain plants, like Ashwagandha (Withania somnifera), Neem (Azadirachta indica), and Giloy (Tinospora cordifolia), are reported in the literature and known to contain bioactive compounds with a wide range of therapeutic benefits like reducing inflammation, lowering fever, antioxidative properties, pain relief, immune system stimulation, and potentially antiviral effects [29-33]. These natural bioactive compounds might help lower mortality rates and block the chain of coronavirus infection. The aim of this study is to deliver a quick overview of the SARS-CoV-2 virus's therapeutic target sites. In addition, the research is aimed at exploring the potential efficacy of these natural chemicals as an alternative therapeutic agent through employing advanced computer-based techniques such as molecular docking and in silico high-throughput screening.
2. MATERIALS AND METHODS
2.1. Retrieval, Preparation and Optimization of Receptor Molecule Spike (S) Protein
The three-dimensional structure (3D) of the spike (S) protein (PDB-ID 7BZ5) [34], a key virulence factor of SARS-CoV-2, was retrieved from the RCSB Protein Data Bank (PDB). To ensure its relevance as a receptor molecule, the spike (S) protein underwent preparation, including the addition of charges and polar hydrogen atoms. Moreover, we marked a grid box to find potential binding sites on the protein. The receptor-binding site was specifically marked and secured at designated coordinates: x (79.684), y (18.727), and z (22.534), employing the Autodock Vina tool [35]. The prepared receptor molecule (spike (S) protein), along with its specified binding site, was then saved in the pdbqt file format for further molecular docking experiments.
2.2. Retrieval, Preparation, and Optimization of Selected Ligand Molecules Derived from Plants
We retrieved the ligand molecules, which are basically bioactive compounds and FDA-approved drugs, from reliable sources like the PubChem Database. Specifically, we selected commonly used drugs in treating SARS-CoV-2, like Remdesivir (PubChem-ID: 121304016), EIDD-2801 (PubChem-ID: 145996610), Dexamethasone (PubChem-ID: 5743), Ritonavir (PubChem-ID: 392622), Hydroxychloro- quine (PubChem-ID: 3652), Favipiravir (PubChem-ID: 492405), and Chloroquine (PubChem-ID: 2711). We got these from the PubChem database (https://pubchem.ncbi. nlm.nih.gov/). However, there was a slight setback because the SDF file format they were in could not be directly used with the Autodock Vina tool [36]. Therefore, we needed to change these SDF files into the PDB file format using the Open Babel GUI [37]. Subsequently, we further converted the PDB files into PDBQT file format and saved them for further in silico experimental analysis using the Autodock Vina bioinformatics software.
2.3. In Silico Parameters Applied for Molecular Docking
We used Autodock Vina for the molecular docking analysis of the screened bioactive compounds and selected FDA-approved drugs [38-40]. The outcomes of the docking indicate how these drugs mentioned above and compounds interact with the structural spike (S) protein of SARS-CoV-2 and exhibited their orientation patterns based on the calculated free binding energy scores in kcal/mol. From these results, we selected the best conformations that had the most favourable binding pocket sites for both drugs and bioactive compounds. Furthermore, these selected conformations were then examined more methodically and used Discovery Studio Visualizer version 2.5.5 and PyMol to find the best patterns in the docked structures. Additionally, we ran the screened bioactive compounds through the Lipinski rule of five (RO5), an assessment of their drug-like properties, using an online server [41]. Further, assessing how effectively these compounds qualified the RO5 prerequisites, our study has provided additional insights into their potential for anti-viral drug development.
2.4. Evaluating the Physiochemical Properties and Potential Toxicity of Screened Bioactive Compounds
In order to predict the ADMET characteristics of the top five bioactive compounds, the study examined their binding affinities [42].
2.5. Molecular Dynamics Simulation (MD Simulation) and In Silico Methodology of Screened Bioactive Compounds
To investigate the stability and flexibility of potent inhibitor bioactive compounds, particularly Withanolide B and Withanolide A, in how they interact with the spike (S) protein, generating a protein-ligand complex. For these simulations, MD simulation and GROMACS v2020 were employedand performed for 100 nanoseconds (100 ns) [43]. Employing MD simulations, researchers may acquire knowledge about the behaviour of biomolecules and the fundamental molecular interactions that impact their dynamics. The ligand compounds were developed by using a simple point charge (SPC) model in a triclinic box topology with the help of the ACPYPE web server [44]. Furthermore, to model how the spike (S) protein receptor acts, we used the CHARMM 27 force field [45], employing a dodecahedron as the simulation box and the TIP3P model for water. Throughout the simulations, we observed the root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) values of the backbone atoms in both the spike (S) protein and the protein-ligand complex. In order to better understand the dynamic behaviour of tested biomolecules, furthermore, we evaluated solvent-accessible surface area (SASA), the radius of gyration (RG), and hydrogen bond (HB) formation.
3. RESULTS
3.1. In Silico Analysis of Certain FDA-approved Drugs and Screened Bioactive Compounds
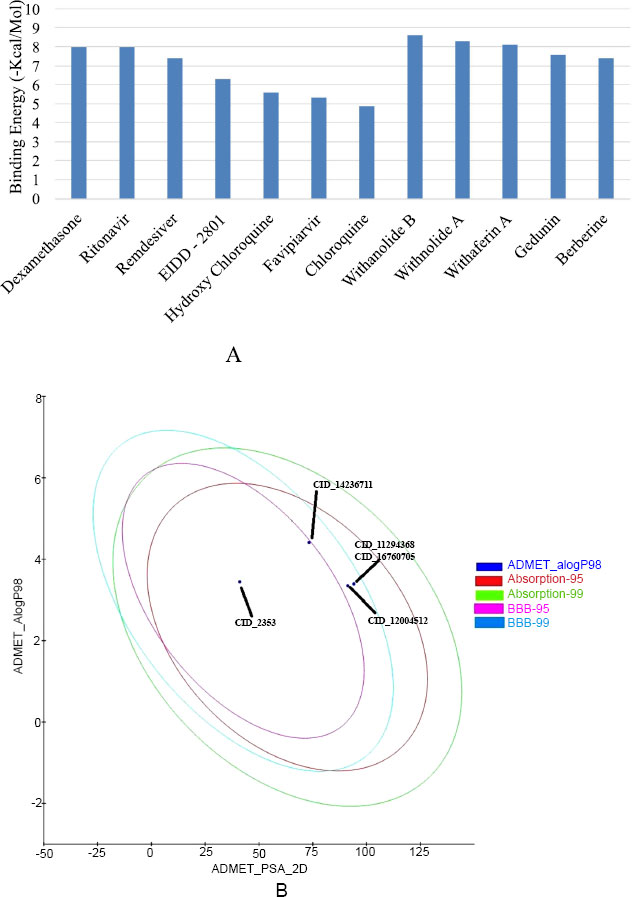
In silico investigation intensive on the SARS-CoV-2 host receptor recognition spike (S) protein, which is essential for the virus's ability to become pathogenic by binding to and fusing with the host cell receptor ACE2. The results of the molecular docking analysis revealed insights into the binding affinities and molecular interaction patterns of screened bioactive compounds with the spike (S) protein. This study especially examined drugs that have been approved by the FDA, such as EIDD-2801, Favipiravir, Ritonavir, Dexamethasone, Chloroquine, and Hydroxychloroquine (Table 1). Apart from that, the plant-derived bioactive compounds, including Withanolide B, Withanolide A, Withaferin A, Gedunin, and Berberine, performed docking with selected receptor molecule spike (S) protein (Table 2). To evaluate the ligands' affinity for the spike (S) protein, we assessed docking energies and scores. Interestingly, the bioactive compounds exhibited higher docked binding energies than the drugs approved by the FDA. Withanolide B, at -8.6 kcal/mol, had the highest docked binding energy, followed by Withanolide A at -8.3 kcal/mol and Withaferin A at -8.1 kcal/mol, respectively. These findings indicate that the bioactive compounds from Withania somnifera that have been evaluated showed excellent possibility of binding to the spike (S) protein's active site. Furthermore, the results validated and compared the molecular docking energies scores of docked bioactive compounds with the spike (S) protein to Dexamethasone, one of the most effective binding drugs (with a free binding energy of -8.0 kcal/mol), preferred as a positive control.
| Sr. No. | Drugs Name | Pub Chem ID |
Binding Energy (Kcal/Mol) |
No. of HB with Amino Acids | No. Hydrophobic Bond | Common Side Effects |
|---|---|---|---|---|---|---|
| 1 | Dexamethasone (C22H29FO5) |
5743 | -8.0 | Ser 373, Ser 371 | Asp 364, Val:362 Val:367 | Adverse effects involving the hair, nails or skin |
| 2 | Ritonavir (C27H35N6O8P) |
392622 | -8.0 | Val:367, Phe:338 gly:339 | Leu 335 | Gastrointestinal adverse effects like diarrhoea, nausea and vomiting |
| 3 | Remdesiver (C27H35N6O8P) |
121304016 | -7.4 | ASN:343, ASN:440, SER:371, SER:373 | TRP:436, PHE:342, LEU:368, LEU 441 | Respiratory, Cardiovascular, Renal toxicity and Hepatotoxicity, |
| 4 | EIDD-2801 (C13H19N3O7) |
145996610 | -6.3 | GLY:339, PHE: 342, ASN:343, SER373, SER371 ,TRP:436,LEU 368 | VAL 367, PHE 338, PHE:374 | Adverse effects on vital organs |
| 5 | Hydroxychloroquine (C18H26ClN3O) |
3652 | -5.6 | ASN:343, TRP:436, ASN:440SER:371 | LEU :368, PHE:342, PHE:342LEU:441 | Hypertension, cataract, neuropsychological disorder and osteoporosis |
| 6 | Favipiravir (C5H4FN3O2) |
492405 | -5.3 | GLU:340, ASN:354, SER:399, ALA 348 PHE347 | VAL:340, ALA:344 | Hyperuricemia and Teratogenicity |
| 7 | Chloroquine (C18H26ClN3) |
2719 | -4.9 | SER 373, SER:371,TRP:436 | PHE :342, PHE:374, LEU:368 | Blurred vision, paresthesia, insomnia |
|
Sr. No. |
Bioactive Compounds | Plant Sources | Pub Chem ID |
Binding Energy (Kcal/Mol) |
No. of HB with Amino Acids | No. Hydrophobic Bond | Common Amino Acid Residue |
|---|---|---|---|---|---|---|---|
| 1 | Withanolide B (C28H38O5) |
Withania somnifera (Ashwagandh) |
14236711 | -8.6 | ASN:343, SER:373 | LEU:441, PHE:374TRP:436 | Phe |
| 2 | Withanolide A (C28H38O6) | 11294368 | -8.3 | CYS:336, GLY:339,SER:373 | LEU:335, PHE:342, TRP:436,PHE:374 | ||
| 3 | Withaferin A (C28H38O6) |
16760705 | -8.1 | SER:373, ASN,440 | TRP:436, PHE:374LEU:441 | ||
| 4 | Gedunin (C28H34O7) |
Azadirachta indica (Neem) |
12004512 | -7.6 | SER:371 SER373PHE 342 | TRP:436 | |
| 5 | Berberine (C20H18NO4+) |
Tinospora cordifolia (Giloy) |
2353 | -7.4 | SER:371, SER:373,CYC 336,GLY 339 | PHE:374, TRP:436VAL:367 |
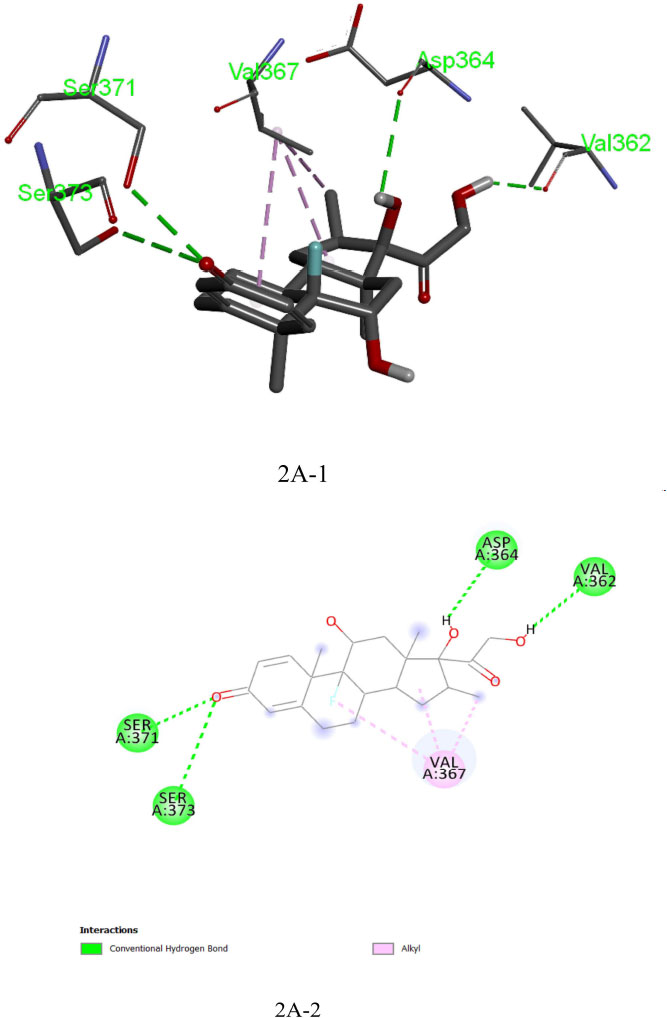
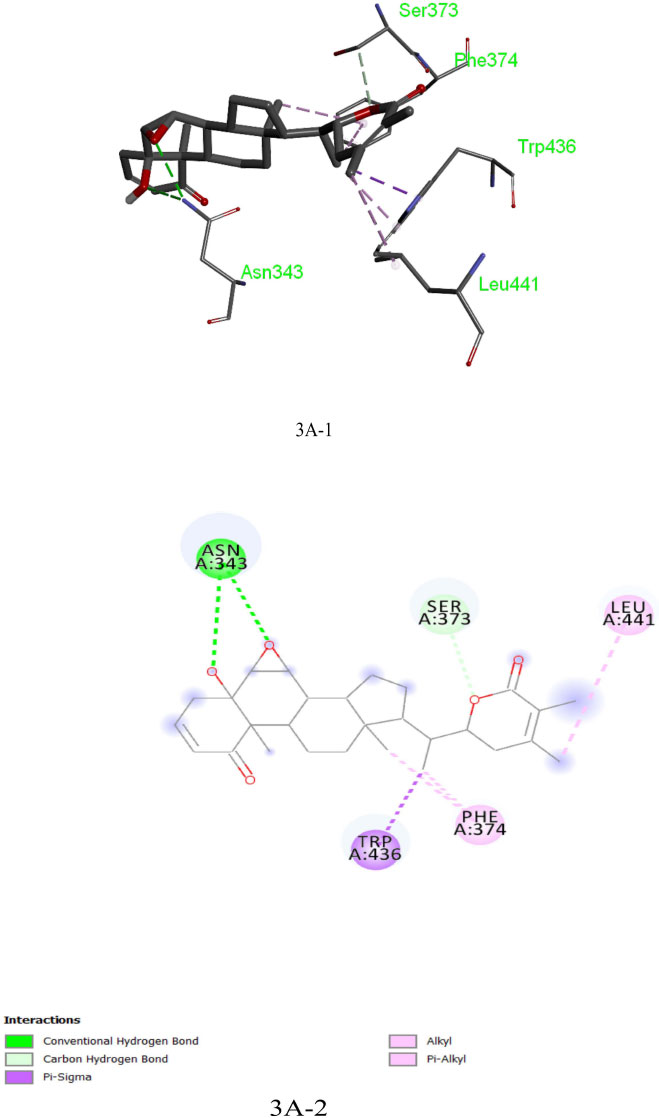
The analysis of Dexamethasone molecular interactions with spike protein revealed hydrogen bonds and hydrophobic bonds with specific amino acid residues, like Ser 373, Ser 371, Asp 364, Val:362 and Val:367 (Fig: 2A-1, 2A-2). In comparison to the docked free energy scores of drugs against the SARS-CoV-2 spike (S) protein, the molecular docking interactions of the screened bioactive compounds exhibited optimal docked free binding energies. Particularly, bioactive compounds like Withanolide B (CID_14236711), Withanolide A (CID_ 11294368), and Withaferin A (CID_16760705) derived from the plant Withania somnifera exhibited docked binding energies of -8.6 kcal/mol, -8.3 kcal/mol, and -8.1 kcal/mol, respectively. Remarkably, Withanolide B (CID_14236711) showed a high affinity for the active site of the SARS-CoV-2 spike (S) protein, corresponding hydrogen bond and hydrophobic bonds to specific amino acid residues, such as ASN:343, SER:373, LEU:441, PHE:374 and TRP:436 (Figs. 3A-1, 3A-2). This binding affinity proved to be higher compared to the affinity of Withanolide A and Withaferin A (see Fig. 4A).
3.2. Analysis of Physicochemical and Toxicity Properties/parameters of Screened Bioactive Compounds
In silico method to analyze the physicochemical and toxicity properties of the top five bioactive compounds: Withanolide B (CID: 14236711), Withanolide A (CID: 11294368), Withaferin A (CID: 16760705), Gedunin (CID: 12004512), and Berberine (CID: 2353). Subsequently, employing Discovery Studio 4.0, the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of these compounds were estimated (see Table 3). The ADME and toxicity assessments of selected bioactive compounds showed drug-like characteristics that make them suitable for human use. Mutagenicity, CYP2 inhibition activity, blood-brain barrier penetration, and intestinal absorption were included among the variables considered into consideration throughout the evaluation. Notably, Withanolide B (CID: 14236711), Withanolide A (CID: 11294368), Gedunin (CID: 12004512), and Withaferin A (CID: 16760705) exhibited high blood-brain penetration and were non-mutagenic. In contrast, the compound berberine (CID: 2353) exhibited minimum blood-brain penetration, while the selected bioactive compounds demonstrated neither mutagenic nor inhibitory effects on CYP450, with the exception of Berberine. These bioactive compounds were also tested for absorption and mutagenicity, and the results showed that they were highly permeable across cell membranes and exhibited nearly no mutagenicity. These compounds are considered non-carcinogenic and highly suitable for drug development, aligning with the Lipinski rule of five (RO5). The compound Withanolide B (CID: 1423611) possessed an evaluation score of -6.301 for ADMET and solubility potential, while Withanolide A achieved a value of -5.2±8. Importantly, none of the compounds that were evaluated had been found to be considered a risk to human health (Fig. 4A, B).
| Sr. No. |
Compound Name and Pubmed ID |
ADMET Solubility | Absorption Level | ADMET BBB | BBB Level | CYP2 Inhibitor | PPB | PPB | Alog | PSA2D | Mutagencity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Withanolide B CID_1423611 |
-6.301 | 0 | -0.05 | 1 | False | 4.62429 | True | 4.411 | 73.277 | Non-mutagen |
| 2 | Withnolide A CID_11294368 |
-5.242 | 0 | -0.593 | 3 | False | 2.83101 | True | 3.397 | 94.092 | Non-mutagen |
| 3 | Withaferin A CID_16760705 |
-5.124 | 0 | -0.5.96 | 3 | False | 2.68768 | True | 3.388 | 94.092 | Non-mutagen |
| 4 | Gedunin CID_12004512 |
-5.994 | 0 | -0.563 | 3 | False | -1.29319 | True | 3.349 | 91.247 | Non-mutagen |
| 5 | Berberine CID_2353 |
-5.541 | 0 | -0.261 | 1 | True | 6.79215 | True | 3.446 | 41.068 | Mutagen |
3.3. MD Simulation Analysis of the Docked Structure of Withanolide B, and Withanolide A Compounds with a Spike (S) Protein of SARS-CoV-2
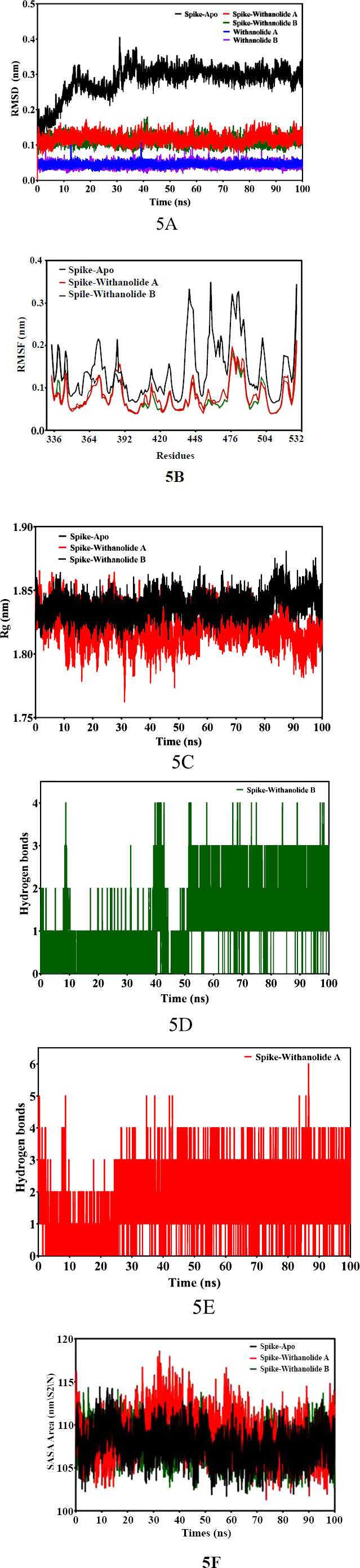
In order to investigate the stability and flexibility of the docked structures of Withanolide B and Withanolide A compounds when combined with the SARS-CoV-2 spike (S) protein, and structural dynamics and the behaviour of protein-ligand complexes were performed MD Simulation [46]. In this study, throughout the simulation process, the docked structures of Withanolide B and Withanolide complexes showed nearly equal affinity binding energy -8.6 kcal/mol and -8.3 kcal/mol, respectively. To assess the stability and rigidity of the docked complexes, we used root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) values obtained during the 100 ns molecular dynamics simulation. The simulation employed a dodecahedron water model (TIP3P). The docked complexes Spike+Withanolide B and Spike+Withanolide A exhibited high stability until the 40 ns time period, while the unbound spike protein of SARS-CoV-2 showed instability with greater variation in the RMSD values and fluctuation (refer to Fig. 5A). Additionally, the fluctuation and compactness of the Withanolide B compound were observed to be high at the 40 ns trajectory time period (refer to Fig. 5B). Consequently, to assess the stability and behaviour of the spike (S) protein of SARS-CoV-2 and its complexes with withanolide B and withanolide A by examining a number of parameters, including radius of gyration (Rg) (refer to Fig. 5C), solvent-accessible surface area (SASA) (refer to Fig. 5F), root mean square deviation (RMSD), root mean square fluctuation (RMSF), and hydrogen bonds (Fig. 5D and 5E) was performed.
4. DISCUSSION
The binding affinities and interaction patterns between the ligands and the receptor protein have been revealed by the molecular docking interactions [47]. The in-silico analysis offers valuable insights into the binding interactions between the spike (S) protein and screened ligands, providing potential therapeutic options for combating SARS-CoV-2. The ADMET analysis revealed favourable properties for the selected bioactive compounds, including Withanolide B, Withanolide A, Withaferin A, Gedunin, and Berberine, in terms of absorption, distribution, metabolism, and excretion. These compounds exhibited high blood-brain penetration, suggesting their potential to reach the central nervous system and exert therapeutic effects. Additionally, the non-mutagenicity and inhibitory activity against CYP450 enzymes in most of the selected bioactive compounds indicate their safety and minimum risk of adverse effects. However, it is important to note that Berberine showed lower blood-brain penetration, suggesting a potential limitation in its ability to reach the central nervous system. The assessment of the compounds' absorption and mutagenicity, along with their adherence to the Lipinski rule of five, further reinforces their potential as drug candidates. These compounds displayed high permeability through cell membranes and exhibited low mutagenicity, making them suitable for consumption and indicating their safety for use as novel drugs. In summary, the in-silico analysis of the physicochemical and toxicity properties of the selected bioactive compounds highlights their promising characteristics as potential therapeutic agents.These findings serve as a foundation for further experimental studies to validate their efficacy and safety profiles. While, the MD simulation analysis provided insights into the stability and flexibility of the docked complexes of Withanolide B and Withanolide A with the spike (S) protein of SARS-CoV-2. The calculated free binding energies demonstrated a strong affinity between the bioactive compounds and the spike protein, indicating their potential as effective binders to the target protein. The docked complexes of Spike+Withanolide B and Spike+Withanolide A preserved their structural integrity and stability for a considerable portion of the simulation time frame in accordance with the stability evaluations conducted via RMSD values. This consistency correlates with findings from other research [48]. The research reports persistent complex forms between the spike protein and bioactive compounds. On the contrary, the unbound spike protein of SARS-CoV-2 displayed greater variability in RMSD values, indicating its inherent flexibility and conformational changes. This finding is consistent with previous studies that have highlighted the dynamic nature of the spike protein in the absence of ligand binding. Moreover, the analysis of fluctuation and compactness in Withanolide B revealed significant changes at specific time points during the simulation. This dynamic behaviour suggests potential conformational adjustments of the compound, which could have implications for its binding interactions and pharmacological effects. Their results, which showed that the bound complexes had lower RMSD values than the unbound protein, were in line with our findings and suggested that the ligand had increased stability. The resemblance points to a widespread pattern of spike protein complexes became more stable. Additionally, studies conducted [49, 50] explored the RMSF patterns of different proteins in order to comprehend their flexibility. They determined that proteins with stable secondary structures had lower RMSF values, which is consistent with our findings. In our study, the bound complexes of Withanolide B and Withanolide A displayed significantly lower RMSF values than the unbound spike protein, indicating a more rigid and stable secondary structure. Regarding hydrogen bonding, our analysis revealed that the bound complex of Withanolide B formed four hydrogen bonds, while the bound complex of Withanolide A formed six hydrogen bonds. This insight correlates with the findings of other studies investigating interactions between proteins and ligands as a case study [51-53], which emphasized the importance of hydrogen bonding in stabilizing protein-ligand complexes. As compared to Withanolide A, Withanolide B's bound complex exhibits fewer hydrogen bonds, which might suggest that the first compound is more stable. Subsequently, to assess protein compactness, comparing the Rg values indicated no significant changes in Rg throughout the simulation, similar findings have been reported in other studies investigating protein dynamics, as seen in a study [54]. The analysis of SASA in this study revealed similar values for the unbound spike protein and the bound complexes of Withanolide B and Withanolide A. This suggests that the binding of these compounds did not significantly impact the folding and unfolding profiles of the protein surface. Therefore, further investigations are necessary to provide a more comprehensive understanding of the impact of ligand binding on protein surface accessibility and thoroughly explore the therapeutic potential of Withanolide B and Withanolide A against the virus.
CONCLUSION
This in silico study highlights the anti-viral potential of Withanolide B and Withanolide A and suggests their role in developing therapeutic agents against SARS-CoV-2 and other viral infections. These bioactive compounds demonstrate significant potential in inhibiting the activity of the spike (S) protein and disrupting the molecular interaction between the viral receptor and host recognition. Their scaffolds hold promise for future therapeutic agents against SARS-CoV-2 and other viral infections, emphasizing the importance of exploring alternative treatments, especially natural bioactive compounds, in the context of the COVID-19 pandemic. Additionally, experimental studies are necessary to establish their efficacy and safety compared to commonly used drugs during the COVID-19 pandemic. These bioactive compounds present a promising avenue for developing novel antiviral treatments, holding significant potential in addressing the ongoing challenges posed by SARS-CoV-2 and other viral pathogens.
LIST OF ABBREVIATIONS
| CSS | = Cytokine storm syndrome |
| ARDS | = Acute respiratory distress syndrome |
| SASA | = Solvent accessible surface area |
| RMSD | = Root mean square deviation |
| RMSF | = Root mean square fluctuation |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIAL
All the data and supporting information are provided within the article.
FUNDING
None.
ACKNOWLEDGEMENTS
The authors express their gratitude to Integral University, Lucknow, and the DST-FIST-funded Department of Bioscience for the essential infrastructure and the Integral University Communication Cell (IUCC) for providing the manuscript communication number (IU/R&D/2024-MCN0002430).

