
Identification and “in silico” Structural Analysis of the Glutamine-rich Protein Qrp (YheA) in Staphylococcus Aureus
Authors Info & Affiliations
Abstract
Background:
YlbF and YmcA are two essential proteins for the formation of biofilm, sporulation, and competence in Bacillus subtilis. In these two proteins, a new protein domain called com_ylbF was recently discovered, but its role and protein function has not yet been established.
Objective:
In this study, we identified and performed an “in silico” structural analysis of the YheA protein, another com_ylbF-containing protein, in the opportunistic pathogen Staphylococcus aureus.
Methods:
The search of the yheA gene was performed using BLAST-P and tBLASn algorithms. The three-dimensional (3D) models of YheA, as well as YlbF and YmcA proteins, were built using the I-TASSER and Quark programs. The identification of the native YheA in Staphylococcus aureus was carried out through chromatography using the FPLC system.
Results:
We found that YheA protein is more widely distributed in Gram-positive bacteria than YlbF and YmcA. Two new and important characteristics for YheA and other com_ylbF-containing proteins were found: a highly conserved 3D structure and the presence of a putative conserved motif located in the central region of the domain, which could be involved in its function. Additionally, we established that Staphylococcus aureus expresses YheA protein in both planktonic growth and biofilm. Finally, we suggest renaming YheA as glutamine-rich protein (Qrp) in S. aureus.
Conclusion:
The Grp (YheA), YlbF, and YmcA proteins adopt a highly conserved three-dimensional structure, harboring a protein-specific putative motif within the com_ylbF domain, which possibly favors the interaction with their substrates. Finally, Staphylococcus aureus expresses the Grp (YheA) protein in both planktonic and biofilm growth.
1. INTRODUCTION
Bacillus subtilis is considered to be the best Grampositive bacterium for study due to its complex processes of cellular differentiation and its ability to produce spores and form biofilm. Many cellular processes were first studied in B. subtilis and later in other Gram-positive bacteria such as Staphylococcus aureus. Biofilm formation in B. subtilis is widely considered an extraordinary survival strategy and a large number of genes have been identified that participate in this complex process [1, 2]. Some studies performed on the mutant derivate B. subtilis 168 strain with deficiency in the formation of spores, competency, and ability to gain foreign DNA allowed for the identification of the ylbF gene, which was needed for competence and sporulation [3]. Later, the ylbF gene was also found to be involved in biofilm formation of both the 168 and NCIB 3610 strains [4]. The ylbF gene encodes a 149-amino acid protein, which contains a new domain called com_ylbF that covers almost all of the protein (120 aa). In addition to the com_ylbF domain, the YlbF protein harbors a Cysteine-rich metallothionein-like motif located in the C-terminal extremity. The com_ylbF domain has also been found in other two proteins, YmcA and YheA. Interestingly, mutations in the ymcA gene (that encodes for the YmcA protein) induce a fail in the biofilm formation of the B. subtilis 168 strain [4]. Additionally, there is experimental evidence showing that the YlbF and YmcA proteins form a stable ternary complex with Yaat protein, which is also required for spore and biofilm formation [5]. Currently, there are two hypotheses about the function and participation of this tripartite complex (YlbF-YmcA-Yaat) in the processes of B. subtilis sporulation, competence, and biofilm formation; the first suggests that this complex stimulates the activity of the master response regulator Spo0A, increasing its phosphorylation [5, 6]. When Spo0A is phosphorylated (Spo0A~P), this regulator activates the transcription of genes needed to form biofilm, mature spores, and competence. Spo0A is phosphorylated by a phosphorelay mechanism, which begins with the auto phosphorylation of the histidine kinases KinA, KinB, KinC, and KinD [7], then the phosphate group is transferred to Spo0F, later to the Spo0B, and finally to the Spo0A [8, 9]. The YlbF-YmcA-Yaat complex accelerates the phosphorelay through the interaction with both Spo0F and Spo0B proteins [5, 6].
The second hypothesis indicates that the YlbFYmcA-Yaat complex is not involved in the phosphorelay process, instead participates in SinR mRNA degradation through a putative functional interaction with the endoribonuclease RNase Y [10, 11]. RNase Y, encoded by the rny gene, is involved in the mRNA decay in different Gram-negative bacteria, including B. subtilis. The SinR protein is a transcriptional repressor that, under biofilm-non-inducing conditions, inhibits the trans-cription of several genes related to biofilm formation and competence, such as the eps and tapA operons, and the comK gene [12]. The sinR gene is finely regulated since little change in the SinR mRNA level causes dramatic changes in biofilm formation [13, 14]. Interestingly, SinR mRNA levels are regulated by RNase Y; mutant strains of the rny gene have shown an increase of 1.4-fold in the amount of the SinR mRNA [15]. This same effect has been observed in mutant strains for ylbF, ymcA, and yaat genes, suggesting that the YlbF-YmcA-Yaat complex could interact with the RNase Y to facilitate the decay of SinR and other mRNAs [10, 11].
Staphylococcus aureus is a Gram-positive bacterium that acts as both a commensal organism and an opportunistic pathogen, causing a broad variety of infections in the human, from superficial skin infections to lethal infections such as bacteremia and pneumonia [16]. In addition, S. aureus also has the ability to form biofilm on both biotic and abiotic surfaces, including indwelling medical devices, which have been associated with chronic and recurrent infections of very difficult treatment. In these infections, S. aureus can attach and persist on host tissues, such as bone and heart valves, to cause osteomyelitis and endocarditis, respectively, or on implanted materials, such as catheters, prosthetic joints, and pace makers [17-19]. With respect to com_ylbF proteins, S. aureus possesses the ylbF and ymcA genes (and also the yheA gene, as found by us in the present study), however, until now, the ortholog proteins that participate in the phosphorelay process or SinR have not yet been identified. By contrast, S. aureus possesses the rny gene that encodes RNase Y, which has also been well characterized in different studies [20-22]. A recent study performed by Deloughery et al. showed that the ylbF gene deletion in S. aureus altered RNA processing by RNase Y [11]. Although the YlbF and YmcA proteins have been relatively well studied in B. subtilis, there is currently no information about the genetic characteristics and function of the YheA protein, the participation of the com_ylbF domain in the protein functionality, or the relationship between three-dimensional (3D) structure and function (to date, the 3D structure of the B. subtilis YmcA protein has only been reported in the PDB database, code 2PIH). Here, we show that the yheA gene in S. aureus possesses a moderately different DNA surrounding with respect to B. subtilis, a similar feature found in ylbF and ymcA genes, but different to the yaat gene, which has very conserved surroundings. Additionally, we show that the yheA gene is more widely distributed in other Gram-positive bacteria than ylbF and ymcA. Multiple alignments and the building of 3D models of YheA proteins allowed for the identification of two new aspects of this protein, a highly conserved 3D structure with respect to YmcA and YlbF, and the presence of a putative conserved motif located in the central region of com_ylbF domain, which could be involved in its function. Finally, we also present experimental evidence that S. aureus express YheA protein in both planktonic and biofilm growth.
2. MATERIALS AND METHODS
2.1. Search for yheA, ylbF, and ymcA Genes in Staphylococcus aureus and Other Gram-Positive Bacteria
The sequences of the YheA, YlbF, and YmcA proteins reported in the genome of the Bacillus subtilis strain 168 (GenBank accession number NP_388861.1, NP_3893 82.1 and NP_389584.1, respectively) were used as a reference to find the yheA, ylbF, and ymcA genes in S. aureus USA300 _FPR3757 and NCTC8325 strains using BLAST-P and tBLASn algorithms. In addition, the genetic surroundings of the yheA, ylbF, and ymcA (and yaaT) genes were established and compared with those reported in B. subtilis. In 2017, Tanner et al. performed an interesting phylogenetic analysis of YlbF, YmcA, and Yaat proteins identified in diverse Gram positive bacteria with or without the ability to form spores [6]. We performed this same analysis but included the YheA protein. The sequences were aligned with the Clustal Omega 1.2.1 program (http://www.ebi.ac.uk/Tools/msa/clustalo/) [23] and visualized using Bioedit. The physicochemical char-acteristics of the proteins were predicted using the ProtParam program and a comparative analysis with putative ortholog proteins reported in UniProt database (http://www.uniprot.org/) was performed. The domains, motifs, and membrane-spanning domains were predicted using the BLAST-P, Pfam, Prosite (http:// pfam.xf am.org/), TMpred (https://embnet. vital-it. ch/software/ TMPRED _form.html), TMHMM (http://www .cbs. dtu. dk/services/ TMHMM/), and CCTOP (http:// cctop.en zim.ttk.mta.hu/) programs. The putative Rho termination sequences were identified using the Transterm program, available at http://transterm.cbcb.umd.edu/ [24].
2.2. Prediction of Three-Dimensional Structures of Proteins
The prediction of 3D structures was performed using the I-TASSER (Iterative Threading Assembly Refinement) program, which first identifies structural templates reported in the Protein DataBank (PDB) using the multiple threading approach, with atomic models constructed by iterative template fragment assembly simulations (http:// zhanglab .ccmb.med.umich.edu/ I-TASSER / ) [25]. The 3D prediction was also carried out using the Quark program, which builds the correct protein 3D model “ab initio” from the amino acid sequence only (https:// zhanglab. ccmb. med. umich. edu/ QUARK/). The 3D models obtained of all the proteins were validated through PROCHECK (http://servicesn.mbi.ucla.edu/ PROCHECK/), ERRAT (http:// services. mbi. ucla. edu/ ERRAT/), and ProSA-web (https:// prosa. services. came. sbg.ac.at/ prosa.php) The Ramachandran plots were built using the Rampage (http:// mordred. bioc. cam.ac.uk/~ rapper/ rampage.php) and PROCHECK programs. The superposition of the 3D models was performed using the MatchMaker application, available in the UCSF Chimera program (https://www.cgl.ucsf.edu/chimera/) [26], and taking as a reference the complete structure of YmcA (2PIH) reported in the PDB database.
2.3. Identification of Native YheA Protein in Staphylococcus aureus in Both Planktonic and Biofilm Growth
In order to establish whether S. aureus expresses the YheA protein, a partial purification scheme based on size-exclusion and ionic-exchange chromatography using the FPLC BioLogic DuoFlowTM system (Bio-Rad®) was conducted. As the physicochemical characteristics of the YheA are not established “in vivo”, we proceeded to produce the recombinant YheA (rYheA) protein and to assess its behavior in both the size-exclusion and ionic-exchange chromatography. Initially, the entire yheA gene (not including the stop codon) was amplified by PCR using the primers CGGTTCTCTA GAATGGCAGTAAATTT ATATGA and TCGTGCATG CATGTCAGCGTAAATTTCG TCTAATG containing XbaI and NseI restriction sites, and fresh DNA isolated from the S. aureus USA300-FPR3757 strain. The DNA fragment (342bp) obtained was first cloned into a pGEM-T easy vector and then subcloned into the Champion™ pET303/CT-His (Invitrogen®) vector (pET-yheA). The recombinant pET-yheA plasmid was transformed first into Escherichia coli TOP10 cells, and was then purified and posteriorly transformed into the T7 polymerase expressing E. coli BL21 cells to produce the rYheA-His6. For the expression and purification of rYheA-His6, the transformed E. coli BL21 cells were grown at 37°C in 50 ml of LB medium containing ampicillin (100 µg/ml) until it reached an OD600 value of 0.6 (~3 h), then protein expression was induced with IPTG 1 mM for 3 hours. These cells were harvested by centrifugation (10,000 rpm), resuspended in 1 ml of buffer (Tris-HCl 0.1 mM, pH: 6.8), and lysed by sonication method (Socincs, vibra cell VCX130®). This suspension was clarified by centrifugation and the supernatant was collected. The soluble rYheA protein was purified by affinity chromatography using the Ni-NTA column coupled to the FPLC BioLogic DuoFlowTM system (Bio-Rad®). The clarified supernatant was applied to the Ni-NTA column and then was washed with 10 column volumes of phosphate buffer (NaH2PO4 50 mM, NaCl 0.5 M pH: 8.0) using a flow of 0.5 ml/min. The interaction rYheA-resin was maintained for 30 minutes. Finally, the rYheA protein was eluted with phosphate buffer plus 250 mM imidazole and fractions were visualized by SDS-PAGE.
Posteriorly, 500 µl of rYheA solution was injected into a size-exclusion Superose 6 HR10/30 column (Pharmacia®) coupled to the FPLC system, which was previously equilibrated with 100 ml of buffer Tris-HCl 20 mM pH 7.5. Forty fractions of 1 ml each were recollected at a flow of 0.3 ml/min. The rYheA protein was identified in the fractions 14 and 15 by SDS-PAGE. Then, these two fractions were joined and injected into an anion-exchange UNO Q1 (Biorad®) column (the rYheA protein was not retained in the cation-exchange column) using buffer Tris-HCl 20 mM pH 7.5 and eluted performing a linear gradient with buffer Tris-HCl 20 mM pH 7.5 supplemented with NaCl 1 M. Seventy fractions of 0.5 ml each were collected at a flow of 0.5 ml/ min. The SDS-PAGE analysis shows that the rYheA protein was eluted with a concentration of NaCl 0.38 M (fraction 49). With these conditions established, we proceeded to seek the native YheA protein in S. aureus growing in both planktonic and biofilm states. For planktonic growth: One colony of USA300-FPR3757 strain was grown in 25 ml of Brain Heart Infusion (BHI) broth at 37°C for 10 hours until reaching the stationary phase of growth. The bacteria were harvested by centrifugation (4,000 rpm), the supernatant was discarded, cells were washed twice then resuspended in 5 ml of sonication buffer (Tris-HCl 0.1 mM, pH: 6.8), and finally treated for 1 hour at 37°C with lysostaphin (50 µg/ml) and lysozyme (1 mg/ml) enzymes. After enzymatic digestion, the bacteria were ice-cooled during 15 min and the lysis was completed by sonication (2 min, 30 s pulse, 15 s break, 30% amplitude). For biofilm growth: One colony of USA300-FPR3757 strain was grown in 5 ml of BHI broth at 37°C for 3 hours. From this suspension, a 1:50 dilution (equivalent to 0.5 McFarland standards) was performed in 20 ml of BHI broth supplement with 1% glucose, place into culture polystyrene dishes, and incubated at 37°C for 24 hours. Then, the supernatant was carefully discarded by pipetting; the adherent cells were washed three times with sterile water, then mechanically detached and recollected in a new tube to be lysed as mentioned above.
Five milliliters of the clarified total protein extract obtained from planktonic or biofilm growth were size-exclusion-separated using a Superose 6 column and buffer Tris-HCl 20 mM pH 7.5. Ten independent chromatographic assays of a 500-µl injection volume were made. The fractions 14 and 15 of each experiment were joined and separated through a single anion interchange chromatography assay using the UNO Q1 column. The fractions were applied on the column with multiple injections using buffer Tris-HCl 20 mM pH 7.5 and eluted with buffer Tris-HCl 20 mM-pH 7.5- NaCl 1M. The fractions between 0.35 M and 0.40 M of NaCl concentration (48 to 50) were recollected and concentrated to half its volume with vacuum concentrator. The proteins contained in these fractions were separated by SDS-PAGE, excised from the gel, and identified by MALDI-TOF-TOF mass spectrometry. The staphylococcal proteins were identified from the peptide mass fingerprint generated using the MASCOT program (http:// www.matrixscience.com/) and their genes located into USA300-FPR3757 and NCTC8325 strains by BLAST.
3. RESULTS
3.1. The yheA Gene has a Distinct Genetic Surrounding in Staphylococcus aureus with Respect to Bacillus subtilis
The com_ylbF domain-containing proteins were initially identified in B. subtilis and it has been well established that at the least two of them, YmcA and YlbF, participate in biofilm formation because their null mutants fail to form pellicles at air-liquid interfaces and grow on solid media as smooth, undifferentiated colonies [3-5]. Curiously, the role and impact in biofilm of YheA, another com_ylbF domain-containing protein, is unknown. With our analysis, we managed to identify the yheA gene in S. aureus, which is located in the chro-mosome at different position with respect to the ylbF and ymcA genes. Other genes found in S. aureus were yaat, rny, comK, clpP, clpC, sfrA, sfrB, sfrC, and sfrD. However, we were unable to find the genes involved in the phosphorelay process and some ortholog of the SinR protein, the two possible targets of the YlbF-YmcA-Yaat complex. The yheA gene has a size of 345 nt and encodes a protein composed of 114 amino acids. Of these, 22% and 10% are negatively and positively charged, respectively, and 13.2% are glutamine (Q). YheA has a theoretical molecular weight of 13.3 kDa, an isoelectric point of 4.3, and is exclusively conformed for α-helix without membrane spanning regions. Similar characteristics were found for YlbF and YmcA proteins. These results suggest that YheA, as for YlbF and YmcA, is probably a soluble cyto-plasmic protein in S. aureus.
A genomic comparative analysis of the yheA surroundings showed significant differences between S. aureus and B. subtilis, generated by the differential insertion of other genes (Fig. 1). A similar result was found for the surroundings of the ylbF and ymcA genes. In contrast, the yaat surroundings were highly conserved in the two bacteria, and it was located together with important bacterial genes such as holB and metS, which encode the DNA polymerase III-subunit δ and methionyl-tRNA synthetase, respectively, suggesting that the Yaat protein could be involved in basic biological processes conserved in these two microorganisms. In the case of the yheA gene, an ORF named yheB, located 78 nt upstream, is conserved in the two bacteria. A detailed analysis of sequence allowed for the identification of a putative Pribnow box and one ribosome binding sequence (AAGGAGT) upstream of the yheB gene (located to 36 and 7 nt upstream of the gene, respectively) (Fig. 1). By contrast, there was a ribosome binding sequence (AAGGAGT) upstream of the yheA gene, but the Pribnow box was not found. Two putative Rho-independent termination sequences located downstream of the yheA were found. However, the global transcription analysis performed in the NCTC8325 strain using RNA-seq has shown that the yheA and yheB genes seem to be independently transcribed (unpublished data, Dr Iñigo Lasa, personal communication). The YheB protein is composed of 374 amino acids and contains two putative membrane-spanning regions located at the residues 24 to 26 and 351 to 373, respectively. This indicates that it could be an anchored membrane protein such as RNase Y.
A phylogenetic analysis performed by Tanner et al. included 28 bacterial species belonging to phylum Firmicutes and showed that the YlbF and YmcA proteins are not exclusive to Bacillus spp. or spore-forming bacteria because there are also found in Staphylococcus spp., Listeria spp., and some species of Lactobacillus spp. and Streptococcus spp [6]. In our analysis, we found a wider distribution of YheA ortholog proteins in the phylum Firmicutes than for YlbF and YmcA because it was identified in all species included in the study (e.g. Pediococcus pentosaceus, Lactobacillus acidophilus, Leuconostoc mesenteroides, Streptococcus pneumoniae, Listeria monocytogenes, and Geobacillus kaustophilus), except for in Clostridia, with identities between 26% (Streptococcus pneumoniae) and 83% (Staphylococcus saprophyticus) (Table 1). This result indicates that the YheA protein could be implicated in basic cellular processes beyond the ability to produce endospores or to form biofilm.
3.2. The YheA protein has a three-dimensional structure that is highly conserved with YmcA and YlbF proteins
The identity percentages of the YheA proteins in different species varied from 26% to 83% (for Streptococcus pneumoniae and Staphylococcus saprophyticus, respectively). To establish the localization of these identical amino acids, we performed a multiple alignment of the YheA protein sequences using Clustal Omega program. Sixteen amino acids were conserved at specific positions along the YheA protein and, interestingly, the QQKQMXG sequence (where X corresponds to an arbitrary amino acid) was found to be conserved and located in the central region in almost all the proteins (Fig. 2a). Based on this finding, we carried out the same analysis for the YmcA and YlbF proteins. In the first analysis, the QKQAVN sequence was located at the residues 50 to 56 and was found to be highly conserved in all YmcA proteins; and in the YlbF analysis, two conserved sequences were also identified, FGXYHPDY and DLNEXV (Fig. 2b). A striking feature that caught our attention was the presence of the amino acid glutamine (Q) in the short conserved sequences of YheA and YmcA. An additional analysis showed that these two proteins have an unusually high content of glutamine, 13.2% and 13.3%, respectively.

To decipher the possible function of YheA, we built and refined a model for the 3D structure of the complete protein employing two approaches; the first by multiple threading from previously reported 3D structures, and the second by “ab initio” modeling, using the I-TASSER and Quark programs. The quality parameters of the models generated for YheA and other proteins are shown in Table 2. The 3D structure of YheA protein is exclusively composed of α-helices (five) (as it was predicted by secondary structure analysis), which are joined for four turns of different lengths (Fig. 3). A similar α-helix structure was obtained when Quark program was used. The α-helices 3 and 4 (residues 36-58 and 64-79, respectively) adopt a spatial arrangement separated from the remaining α-helices in the structure, forming an acute angle, where a conserved aspartic acid at the position 35 acts as a hinge. In addition, these α-helices are linked by a long turn composed of five amino acids (GEEIA). The entire 3D structure of YmcA has been experimentally established (PDB code: 2PIH), showing that this protein is a homodimer, which has also been confirmed in in vitro using FPLC analysis with the recom-binant protein [5]. From these results, we built the models as homodimers (Fig. 3). Notably, the conserved QQKQMXG sequence observed in almost all YheA proteins is located within the third α-helix. We extended the structural modeling to YmcA and YlbF of S. aureus and YheA and YlbF of B. subtilis (Fig. 3). However, we came across a difficulty in modeling the complete YlbF protein of these microorganisms, since all models generated the Cysteine-rich metallothionein like motif (composed of approx. 17 amino acids and located in the C-terminal extremity) adopting a disordered and highly variable structure. Nevertheless, the com_ylbF domain had a conformation with five α-helices, with a similar 3D arrangement with respect to the YheA and YmcA proteins.
| % Identity | |||||
|---|---|---|---|---|---|
| Bacteria | YheA | YlbF | YmcA | ||
| Bacilli | Lactobacillales | Pediococcus pentosaceus | 48% | - | - |
| Lactobacillus plantarum | 43% | - | - | ||
| Lactobacillus fermentum | 44% | - | - | ||
| Leuconostoc mesenteroides | 36% | - | - | ||
| Lactobacillus acidophilus | 31% | - | - | ||
| Streptococcus sanguinis | 30% | - | - | ||
| Streptococcus pneumoniae | 26% | - | - | ||
| Streptococcus salivarius | 33% | - | - | ||
| Streptococcus mutans | 34% | 20% | 24% | ||
| Lactobacillus delbrueckii | 32% | 30% | 14% | ||
| Non-sporulating Bacillales | Enterococcus faecalis | 48% | 22% | 34% | |
| Listeria monocytogenes | 40% | 29% | 44% | ||
| Staphylococcus aureus | 100% | 100% | 100% | ||
| Staphylococcus saprophyticus | 83% | 54% | 69% | ||
| Sporulating Bacillales | Bacillus anthracis | 47% | 34% | 42% | |
| Bacillus subtilis | 47% | 31% | 40% | ||
| Bacillus halodurans | 45% | 31% | 39% | ||
| Oceanobacillus iheyensis | 47% | 30% | 38% | ||
| Exiguobacterium sibiricum | 46% | 32% | 35% | ||
| Lysinibacillus sphaericus | 42% | 37% | 40% | ||
| Geobacillus kaustophilus | 33% | 38% | 45% | ||
| Clostridia | Sporulating Clostridia | Alkaliphilus metalliredigens | - | - | - |
| Clostridium botulinum | - | - | - | ||
| Natranaerobius thermophilus | - | - | - | ||
| Heliobacterium modestica | - | - | - | ||
| Desulfitobacterium hafniense | - | - | - | ||
| Carboxydothermus hydrogenoformans | - | - | - | ||
| Syntrophomonas wolfei | - | - | - | ||
Therefore, we performed a structural comparative analysis with the 3D structure model obtained from partial YlbF sequence (residues 1-128, without the metallothionein-like motif). Interestingly, the YheA, YmcA, and YlbF proteins possess a highly conserved 3D structure, composed mainly of five α-helices, two of which (α-helices 3 and 4) form a <90° angle with the three remaining α-helices. In the YheA protein, this angle is lower compared to YlbF and YmcA. Additionally, the short conserved sequences found in both the YmcA and YlbF proteins (QKQAVN and FGXYHPDY, respectively) are also located within the third α-helix, as occurs in the YheA protein. When the homodimers of the three proteins are built, they adopt a shape of “pincers” with the short conserved sequences located at their “jaw”, where these amino acids could exert their function.


| - | S. aureus | B. subtilis | ||||
|---|---|---|---|---|---|---|
| - | YheA | YmcA | YlbF** | YheA | YmcA* | YlbF** |
| Ramachandran plot | ||||||
| Residues in most favored regions | 98.2% | 92.3% | 93.1% | 96.4% | 97.5% | 91.4% |
| Residues in additional allowed regions | 0.9% | 5.1% | 6.0% | 3.6% | 2.5% | 7.8% |
| Residues in generously allowed regions | 0.9% | 1.7% | 0.9% | 0% | 0% | 0% |
| Residues in disallowed regions (outlier) | 0% | 0.9% | 0% | 0% | 0% | 0.9% |
| Overall G-factor | 0.01 | - 0.07 | - 0.08 | - 0.08 | 0.56 | - 0.11 |
| Z-score | - 4.52 | - 4.98 | - 3.99 | - 4.21 | - 4.07 | - 3.87 |
| C-score | 0.46 | 1.16 | 0.56 | 0.40 | - | 0.54 |
| TM-score | 0.77 | 0.87 | 0.79 | 0.77 | - | 0.79 |
| RMSD | 3.3 | 2.1 | 3.2 | 3.5 | - | 3.3 |
| ERRAT | 100 | 96.5 | 100 | 100 | 100 | 100 |
3.3. The YheA Protein is Expressed in Staphylococcus Aureus
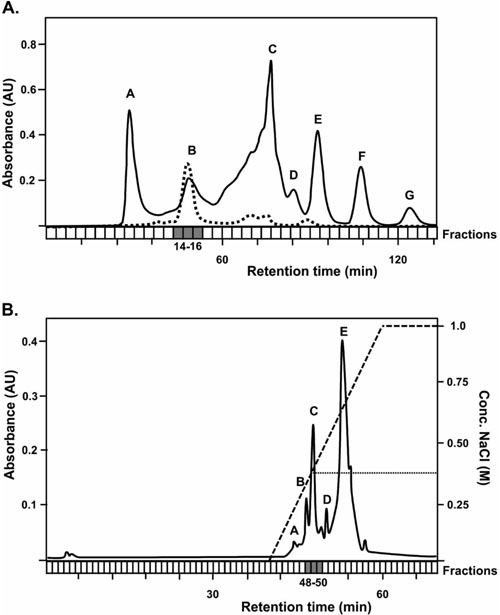
Since there is currently no experimental evidence concerning the expression YheA, we used a partial purification process based on chromatography separations to try to establish whether S. aureus expresses the YheA protein. Firstly, we attempted to detect YheA in the size-exclusion chro-matography fractions 26 to 30 (fractions where the proteins with a molecular weight between 10 to 20 kDa are expected) but it was not successful (Fig. 4). Then, we produced and purified the recombinant YheA protein (rYheA) and assessed its behavior in the size-exclusion chromatography. We found that rYheA protein eluted faster, within fractions 14, 15, and 16 (peak B, Fig. 4) where proteins with a greater molecular weight are expected. Using protein extract of cells growing in late exponential phase and forming biofilm, the native YheA protein was also identified in fractions 14, 15, and 16 (peak B). This result suggests that YheA protein could be forming oligomers or interacting with other proteins forming larger protein complex.
4. DISCUSSION
In 2000, the ylbF protein was identified for the first time in a B. subtilis strain unable to produce spores, acquire foreign DNA, or form biofilm, generated through random mutagenesis experiments [3-5]. The analysis of this protein allowed for the discovery of a new protein domain called com_ylbF, which was composed of 120 amino acids. Currently, two additional proteins that possess this same com_ylbF domain have been reported, YmcA and YheA. It has been established that ymcA deletion causes an abolishment of the ability to form biofilm, spores, or competency [4, 5]. By contrast, very little is known about YheA. Our analysis shows that YheA is more frequently found in the phylum Firmicutes than YlbF and YmcA, even within species without the ability to produce spores (Table 1), indicating that YheA could be involved in some basic biologic process conserved in this phylum. Although there are differences between the genetic surroundings of yheA in S. aureus and B. subtilis, it is always found close to yheB, suggesting that these two genes could form part of an operon. However, preliminary results of S. aureus RNA-seq experiments indicate that they are independently transcribed and possibly regulated by different pathways (unpublished data, Dr Iñigo Lasa, personal communication).
The principal contributions of the present study are: the identification of a highly conserved 3D structure in three com_ylbF domain-containing proteins despite their low amino acid identity (<16%), and the identification of protein-specific short conserved sequences, which could act as new putative motifs in each protein and could be involved in its function. Although the in silico model of proteins can contain errors, they may be useful as they allow for the prediction of possible functions from certain coarse structural features to efficiently direct biological experiments. Currently, two different and apparently contradictory roles have been attributed for YlbF and YmcA proteins (but none for YheA). The first involves facilitating the phosphorylation of transcriptional regulator Spo0A through the formation of two (4Fe-4S)+2 clusters [5, 6, 27], and the second, regulating the mRNA maturation interacting with the endoribonuclease RNase Y [10, 11]. In the first case, the activity of the proteins has only been associated with the metallothionein-like motif but not the com_ylbF domain, and is completely unknown in the second. In conclusion, the more important domain of these proteins has not yet revealed its function. Our results indicate that a function of the com_ylbF domain could exist, which would be mediated by a putative motif composed of three to five highly conserved amino acids and would be specific for each protein. Now, whether the proteins YheA, YmcA, and YlbF act as homodimers, such as has previously been reported for YmcA [5], the putative motifs would be located within two highly conserved α-helices where the amino acids are oriented toward the interior of the protein and where could interact more adequately with their substrates (maybe RNA molecules, due to the RNA-binding ability predicted for YlbF [28]), simulating a “pincer”. This finding requires further sitedirected mutagenesis experiments in YheA (as well as YlbF and YmcA) in both S. aureus and B. subtilis to corroborate the importance of these putative motifs in biofilm formation, competence, and sporulation (the latter in the case of B. subtilis). Additionally, based on the high content of glutamine found in the complete protein and the putative motif, as well as its possible participation in the function of the protein, we suggest renaming YheA as glutamine-rich protein (Qrp) in S. aureus.
Finally, in addition to all the information concerning YlbF and YmcA in B. subtilis, it is important to establish the function of these proteins in other bacteria such as S. aureus, which could provide more evidence to support its participation in either the phosphorelay process or RNA decay. Curiously, S. aureus does not possess proteins related to Spo0A or SinR, but it has an ortholog of RNase Y. However, although it does not seem essential since its deletion does not generate a high genetic cost, its mutants have lower pathogenicity [20-22]. Recently, Deloughery et al. showed that the ylbF gene is required for mRNA cleavage of the cggR-gapA operon by RNase Y in S. aureus [11]. Experimental evidence suggests that RNase Y could be implicated in multiple roles in S. aureus, from RNA decay up to the processing and stabilization of some mRNA transcripts (e.g. the saePQRS operon) [20-22], as it has also been reported in Streptococcus pyogenes [29]. Then, in addition to the com_ylbF proteins being associated with the RNase Y (as happen in B. subtilis), they could be involved in another additional role in S. aureus.
CONCLUSION
Grp (YheA) is another com_ylbF domain-containing protein, which is widely conserved among bacteria with or without the ability to form spores, indicating that its function could be related beyond this process. Bioinformatic analyses suggest that Grp (YheA), YlbF, and YmcA proteins adopt a highly conserved three-dimensional structure, composed exclusively of α-helices, with homodimers spatially arranged to form a “pincer”. In addition, these three proteins seem to harbor a protein-specific putative motif in the com_ylbF domain, located at the extremes of the “pincer”. The strategic and conserved localization of these motifs possibly favors the interaction with their substrates, facilitating the function of the proteins. Finally, Staphylococcus aureus expresses the Grp (YheA) protein in both planktonic and biofilm growth.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
This study was supported by the Departamento Administrativo de Ciencia, Tecnología e Innovación, Colciencias (grant number 1308-657-41107 607-2014) and Vice Chancellery for Research of Universidad El Bosque (grant numbers PCI-2013-373 and PCI2017- 9614). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. We gratefully acknowledge to Dr Miguel Otero for your invaluable support and Dra Ana Lucia Castiblanco for your technical assistance in the FPLC system.

