
Identification and Characterization of Novel Mutants of Nsp13 Protein among Indian SARS-CoV-2 Isolates
Authors Info & Affiliations
Abstract
Background:
SARS-CoV-2, the causative agent of COVID-19, has mutated rapidly, enabling it to adapt and evade the immune system of the host. Emerging SARS-CoV-2 variants with crucial mutations pose a global challenge in the context of therapeutic drugs and vaccines developing globally. There are currently no specific therapeutics or vaccines available to combat SARS-CoV-2 devastation. Concerning this, the current study aimed to identify and characterize the mutations found in the Nsp13 of SARS-CoV-2 in Indian isolates.
Methods:
In the present study, the Clustal omega tool was used for mutational analysis. The impact of mutations on protein stability, flexibility, and function was predicted using the DynaMut and PROVEAN tools. Furthermore, B-cell epitopes contributed by Nsp13 were identified using various predictive immunoinformatic tools.
Results:
Non-structural protein Nsp13 sequences from Indian isolates were analyzed by comparing them with the firstly reported Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) protein sequence in Wuhan, China. Out of 825 Nsp13 protein sequences, a total of 38 mutations were observed among Indian isolates. Our data showed that mutations in Nsp13 at various positions (H164Y, A237T, T214I, C309Y, S236I, P419S, V305E, G54S, H290Y, P53S, A308Y, and A308Y) have a significant impact on the protein's stability and flexibility. Moreover, the impact of Nsp13 mutations on protein function was predicted based on the PROVEAN score that indicated 15 mutants as neutral and 23 mutants as deleterious effects. Immunological parameters of Nsp13, such as antigenicity, allergenicity, and toxicity, were evaluated to predict the potential B-cell epitopes. The predicted peptide sequences were correlated with the observed mutants. Our predicted data showed that there are seven high-rank linear epitopes as well as 18 discontinuous B-cell epitopes based on immunoinformatic tools. Moreover, it was observed that out of the total 38 identified mutations among Indian SARS-CoV-2 Nsp13 protein, four mutant residues at positions 142 (E142), 245 (H245), 247 (V247), and 419 (P419) were localised in the predicted B cell epitopic region.
Conclusion:
Altogether, the results of the present in silico study might help to understand the impact of the identified mutations in Nsp13 protein on its stability, flexibility, and function.
1. INTRODUCTION
The recent emergence of an outburst of coronavirus disease 2019 (COVID-19) pandemic caused by a novel beta coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1], was first reported in Wuhan, China, in late December 2019 [2-5]. COVID-19 spread rapidly worldwide and was declared a global pandemic on 11th March, 2020, by World Health Organization (WHO). It has infected hundreds of millions of people and killed more than three million people worldwide. The world is in the midst of the second wave of a pandemic with rapidly evolving new variants of SARS-CoV-2, resulting in a surge of COVID-19 cases worldwide. The highly contagious nature of SARS-CoV-2 has now developed into a severe threat to global public health. The manifestations of SARS-CoV-2 range from the asymptomatic common cold to lethal viral respiratory illness in infected individuals [6].
Coronaviruses are enveloped in a nonfragmented, large single-stranded (positive sense) RNA virus with a GC content ranging from 32% to 43%. They are classified into four genera, namely, alphacoronavirus (α CoV), beta coronavirus (β CoV), gamma coronavirus (γ CoV), and delta coronavirus (δ CoV), respectively [7]. SARS-COV-2 belongs to the β coronavirus 2b lineage with a genome of length 29.9 kb, which encodes 29 proteins [8]. The genomic sequence of the novel SARS-COV-2 exhibits 79.6% similarity to SARS-CoV and about 50% with MERS-CoV (Middle East respiratory syndrome coronavirus), respectively. The phylogenetic analysis has illustrated that SARS-CoV-2 is relatively more similar to SARS-CoV than MERS-CoV. Based on homology modelling studies, SARS-CoV-2 shares 96.2% nucleotide similarity with RaTG13, a bat CoV from Rhinolophus Affinis. The genomic organisation of SARS-CoV-2 includes '5 UTR, ORF1ab, S, E, M, N, and 3'UTR. Its genome comprises ORFs, which encode for four major structural proteins, Spike glycoprotein (S), Envelope (E), Membrane (M), and Nucleocapsid (N) proteins, 16 Nsps (non-structural proteins), and nine other accessory proteins [9-11]. ORF1a/b region is the largest ORF that covers about two-thirds of the whole genome length and encodes known sixteen non-structural proteins (16Nsps). ORFs in the remaining one-third of the genome near the 3' end code for major structural and other accessory proteins. The ribosomal frameshift (-1) between ORF1a and ORF1b produces two polypeptides (pp1a and pp1ab), which are proteolytically cleaved by viral proteases, namely, main protease (M pro) or Chymotrypsin-like protease (3CLpro) and papain-like protease (PL pro) to release 16 Nsps of 7096 amino acids in length. These Nsps are crucially important in viral replication and transcription processes [8]. Nsps are considered more conserved than SARS-CoV-2 structural proteins [12]. Among the 16 known SARS-CoV-2 Nsp proteins, the Nsp13 helicase is a crucial component for viral replication [13] and the most conserved nonstructural protein with 99.8% sequence identity to SARS-CoV Nsp13 [14]. The highest sequence conservation is shared by SARS-CoV-2 Nsp13 within the CoV family, reflecting its significant role in viral viability. Nsp13, an RNA helicase enzyme, also represents a promising therapeutic target for anti-CoV drug development [15-18].
The ongoing rapid transmission and global spread of SARS-CoV-2 have acquired various mutations. The global advancements in the sequencing efforts of SARS-CoV-2 have reported several mutations in its proteins, like spike protein, nucleocapsid, PLpro, and ORF3a, from different populations [19-21]. Emerging variants of SARS-CoV-2 with crucial mutations is a sparkling challenge in the context of therapeutics, drugs, and vaccines developing globally. To date, there are no specific therapeutics and vaccines available to combat SARS-CoV-2. Given this, the present study aimed at identifying and characterizing mutations in SARS-CoV-2 Nsp13 protein that might prove to be useful towards the development of drugs and effective vaccines against this highly mutable coronavirus. It could also help in improving the current diagnostic approaches to viral detection, thereby controlling the transmission of the virus. As a consequence of the indispensable role of SARS-Cov-2 Nsp13 in the replication of the virus, the study aimed at understanding the possible structural and functional implications of Nsp13 mutations among Indian isolates.
2. METHODS
2.1. Sequence Retrieval and Multiple Sequence Alignment
As of 24th May, 2021, there were 825 ORF1ab protein sequences of Indian SARS-CoV-2 deposited in the NCBI Virus database. All these ORF1ab protein sequences (7096 residues) comprising nonstructural proteins (Nsp1-16) were retrieved from the NCBI-Virus database. The protein accession numbers of ORF1ab sequences used in the study are mentioned in the Table S1. The first reported sequence from China was used as a reference or wild type ORF1ab sequence (accession number: YP_009724389) for mutational analysis in the study. The polypeptide sequences of ORF1ab were exported in the FASTA format, in which the Nsp13 of 601 residues starting from 5325 to 5925 were extracted from the ORF1ab sequence. Nsp13 is proteolytically cleaved by the protease encoded by the SARS-CoV-2 genome. The Clustal Omega online web server tool was used for multiple sequence alignment (MSA) [22, 23] to identify mutations in the amino acid sequences of Nsp13 among the Indian isolates. Clustal Omega is a widely used online program for efficiently generating alignments using a fast and reliable algorithm. For this analysis, the Indian sequences were compared with the reference sequence of Nsp13 (accession number: YP_009724389), and then the identified mutant residues were marked carefully and noted for further analysis.
2.2. Prediction of the Effect of Mutations on Protein Stability and Dynamicity
To understand the impact of mutational changes on the protein stability and dynamicity of Nsp13, protein modelling studies were performed by using DynaMut, a web server tool [24]. The 3D structure of the target protein Nsp13 with PDB ID-6ZSL was used for protein modelling studies. DynaMut program is widely used to analyse and visualise the changes in protein dynamics and stability caused by mutations in terms of the difference in vibrational entropy (∆∆S) and free energy (∆∆G) between the wild type and mutant proteins. The effect of mutations on changes in protein stability was estimated by the predicted ∆∆G(Kcal/mol) between wild-type and mutant proteins. The positive values (more than zero) indicated the stabilization of protein structure and negative values (below zero) showed the destabilization of the protein structure. The protein flexibility and rigidity were calculated by the difference in vibrational entropy (∆∆S) between wild-type and mutant proteins of Nsp13. The positive ∆∆S values corresponded to an increase in the flexibility of the Nsp13 protein, whereas the negative ∆∆S values represented a decrease in the flexibility of the protein.
2.3. Prediction of PROVEAN Score
To understand the effect of amino acid variations on the function of Nsp13, PROVEAN (Protein Variation Effect Analyzer) tool was used [25]. This prediction tool was used to generate a PROVEAN score for each variant. PROVEAN provides an approach to predicting the functionally important sequence variants. Based on the predicted PROVEAN score, mutants are classified as deleterious or neutral. The protein variant is predicted to have a deleterious effect if the PROVEAN score is equal to or less than the default threshold score (-2.5) and if the PROVEAN score is more than the threshold score, the variant is predicted to have a neutral effect. PROVEAN tool is useful in measuring the functional effect of protein sequence variations, including amino acid substitution, insertion, and deletions.
2.4. Prediction of B-cell Epitopes for Nsp13
B-cell epitope predictions were performed using an online web server IEDB (Immune Epitope Database) prediction tool [26], as described by Jesperson et al. [27]. This prediction tool predicts the linear continuous B-cell epitopes of Nsp13 based on the BepiPred linear epitope prediction method by using all the standard parameters, such as flexibility, hydrophilicity, accessibility, and beta turns at a threshold value of 0.48. The identified epitopes of Nsp13 were further assessed for immunological parameters, such as antigenicity, allergenicity, and toxicity (by Toxin Pred). Antigenicity and allergenicity of peptides were predicted by freely available web servers, VaxiJen (http://www.jenner.ac.uk/VaxiJen) [28] and Allergen FP v.1.0 (http://ddg-pharmfac.net/AllergenFP) [29]. Moreover, the discontinuous B-cell epitopes were also predicted using Discotope 2.0 web server tool [30] at a threshold value of -6.6 followed by representing the identified epitopes in the three-dimensional structure of Nsp13 protein. The Discotope score of residues above the threshold value indicates positive prediction and those below the threshold value indicate negative prediction, respectively.
3. RESULTS
3.1. Identification of Mutations in Nsp13 among Indian SARS-Cov-2 Isolates
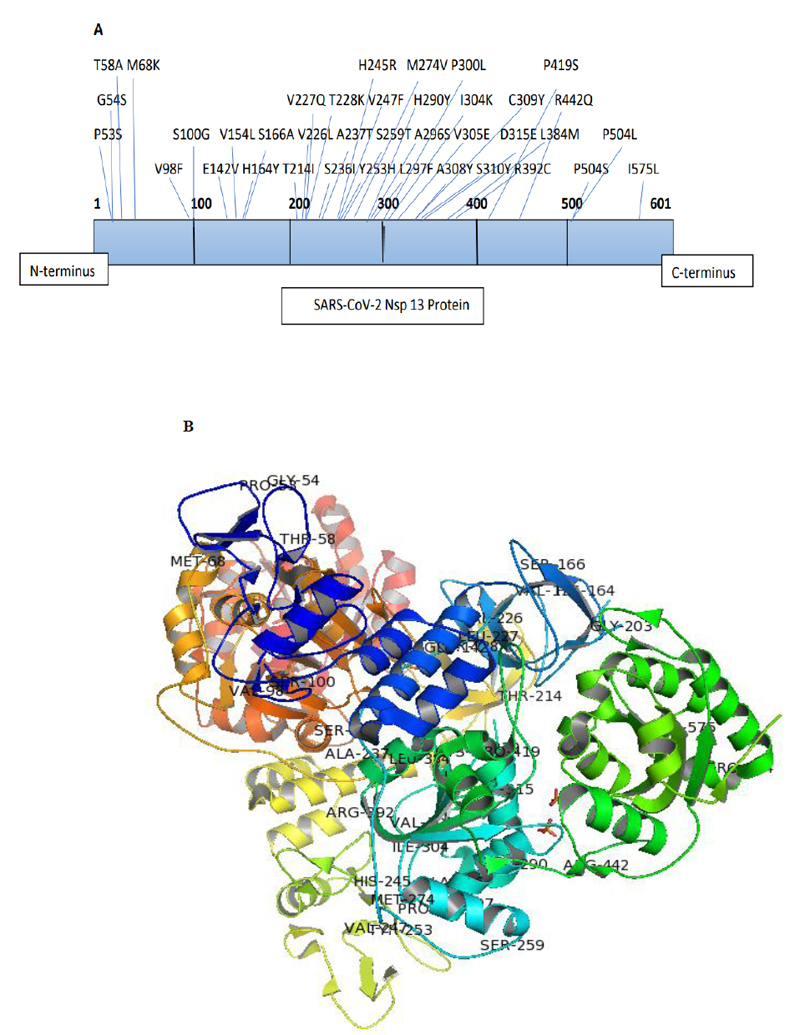
The Nsp13 SARS-CoV-2 sequences reported from India till 24th May, 2021, were aligned with the reference sequence reported from China (accession number: YP_009724389) using Clustal omega-mediated multiple sequence alignment for mutational analysis. The amino acid variations in the Nsp13 protein sequences among Indian isolates were identified carefully and noted for analysis. The analysis revealed the presence of 38 mutations distributed all over the Nsp13 protein. The details of each mutation are listed in Table 1 and their location observed are represented in the three-dimensional structure of Nsp13 (PDB ID-6ZSL) using the Pymol visualisation tool (Fig. 1). Our data also demonstrated a change in polarity and charge upon amino acid mutations in Nsp13. It was found that most of the mutations did not contribute to any changes (Neutral to Neutral). Although some of these mutations led to an alteration in charge from basic to neutral (H164Y, H290Y, R392C, and R442Q), only one mutation at position 142 (H142V) altered the charge from acidic to neutral, as depicted in Table 1.
| S. No. | Mutation | Polarity Changes | Charge Changes |
|---|---|---|---|
| 1 | P53S | NP to P | Neutral to Neutral |
| 2 | G54S | P to P | Neutral to Neutral |
| 3 | T58A | P to NP | Neutral to Neutral |
| 4 | M68K | NP to P | Neutral to Basic |
| 5 | V98F | NP to NP | Neutral to Neutral |
| 6 | S100G | P to P | Neutral to Neutral |
| 7 | E142V | P to NP | Acidic to Neutral |
| 8 | V154L | NP to NP | Neutral to Neutral |
| 9 | H164Y | P to P | Basic to Neutral |
| 10 | S166A | P to NP | Neutral to Neutral |
| 11 | T214I | P to NP | Neutral to Neutral |
| 12 | V226L | NP to NP | Neutral to Neutral |
| 13 | L227Q | NP to P | Neutral to Neutral |
| 14 | T228K | P to P | Neutral to Basic |
| 15 | S236I | P to NP | Neutral to Neutral |
| 16 | A237T | NP to P | Neutral to Neutral |
| 17 | H245R | P to P | Basic to Basic |
| 18 | V247F | NP to NP | Neutral to Neutral |
| 19 | Y253H | P to P | Neutral to Basic |
| 20 | S259T | P to P | Neutral to Neutral |
| 21 | M274V | NP to NP | Neutral to Neutral |
| 22 | H290Y | P to P | Basic to Neutral |
| 23 | A296S | NP to P | Neutral to Neutral |
| 24 | L297F | NP to NP | Neutral to Neutral |
| 25 | P300L | NP to NP | Neutral to Neutral |
| 26 | I304K | NP to P | Neutral to Basic |
| 27 | V305E | NP to P | Neutral to Acidic |
| 28 | A308Y | NP to P | Neutral to Neutral |
| 29 | C309Y | P to P | Neutral to Neutral |
| 30 | S310Y | P to P | Neutral to Neutral |
| 31 | D315E | P to P | Acidic to Acidic |
| 32 | L384M | NP to NP | Neutral to Neutral |
| 33 | R392C | P to P | Basic to Neutral |
| 34 | P419S | NP to P | Neutral to Neutral |
| 35 | R442Q | P to P | Basic to Neutral |
| 36 | P504S | NP to P | Neutral to Neutral |
| 37 | P504L | NP to NP | Neutral to Neutral |
| 38 | I575L | NP to NP | Neutral to Neutral |
3.2. Effect of Mutation on Stability and Flexibility
The mutational analysis for evaluating the impact of mutations on the stability and flexibility of Nsp13 protein was conducted by the DynaMut web server. This web server calculated the difference in free energy(∆∆G) between wild-type and mutants. The positive ΔΔG corresponded to an increase in stability, while the negative ΔΔG corresponded to a decrease in stability. Our data demonstrated that a total of 38 mutations caused an increase or decrease in the stability of Nsp13 protein. The positive ∆∆G values in the range of 0.013 to 0.975 kcal/mol showed a stabilising effect (Table 1). We observed the maximum positive ΔΔG for H164Y (0.975), A237T (0.924), T214I (0.881), C309Y (0.844), S236I (0.750), and P419S (0.693), indicating stabilising mutations (Fig. 2). While, V305 (-2.53), G54S (-1.69), and A308Y (-1.306) showed a high negative ∆∆G value, indicating the destabilization effect on the Nsp13 protein (Fig. 2). Similarly, the flexibility was also calculated by the difference in vibrational entropy (∆∆S) between the wild-type and mutant protein of Nsp13. The maximum positive ∆∆S (0.896) for H290Y caused an increase in the flexibility of Nsp13 protein whereas, the maximum negative ∆∆S (-0.725) for A308Y represented a decrease in flexibility of the protein (Table 2). Collectively, our data suggested that mutations identified in the Nsp13 protein could alter its stability and dynamicity.
| S. No | Chain ID | Mutation | Predicted ΔΔG (kcal/mol) | Stability |
Predicted ΔΔSVib ENCoM (kcal. mol -1. K-1) |
Flexibility |
|---|---|---|---|---|---|---|
| 1 | A | P53S | 0.024 | Stabilizing | 0.504 | Increase |
| 2 | A | G54S | -1.694 | Destabilizing | 0.130 | Increase |
| 3 | A | T58A | -0.260 | Destabilizing | 0.358 | Increase |
| 4 | A | M68K | 0.188 | Stabilizing | 0.165 | Increase |
| 5 | B | V98F | 0.160 | Stabilizing | -0.193 | Decrease |
| 6 | B | S100G | -0.157 | Destabilizing | 0.413 | Increase |
| 7 | A | E142V | -0.174 | Destabilizing | 0.260 | Increase |
| 8 | A | V154L | 0.235 | Stabilizing | 0.031 | Increase |
| 9 | A | H164Y | 0.975 | Stabilizing | -0.375 | Decrease |
| 10 | A | S166A | -0.100 | Destabilizing | 0.145 | Increase |
| 11 | A | T214I | 0.881 | Stabilizing | -0.339 | Decrease |
| 12 | A | V226L | 0.528 | Stabilizing | -0.236 | Decrease |
| 13 | A | L227Q | -0.268 | Destabilizing | 0.015 | Increase |
| 14 | A | T228K | 0.525 | Stabilizing | -0.315 | Decrease |
| 15 | A | S236I | 0.750 | Stabilizing | -0.370 | Decrease |
| 16 | A | A237T | 0.924 | Stabilizing | -0.424 | Decrease |
| 17 | A | H245R | -0.154 | Destabilizing | 0.147 | Increase |
| 18 | A | V247F | -0.096 | Destabilizing | -0.102 | Decrease |
| 19 | A | Y253H | 0.309 | Stabilizing | -0.109 | Decrease |
| 20 | A | S259T | 0.031 | Stabilizing | -0.031 | Decrease |
| 21 | A | M274V | -0.690 | Destabilizing | 0.441 | Increase |
| 22 | A | H290Y | 0.562 | Stabilizing | 0.896 | Increase |
| 23 | A | A296S | -1.135 | Destabilizing | -0.128 | Decrease |
| 24 | A | L297F | 0.013 | Stabilizing | -0.252 | Decrease |
| 25 | A | P300L | 0.362 | Stabilizing | -0.427 | Decrease |
| 26 | A | I304K | -0.410 | Destabilizing | -0.022 | Decrease |
| 27 | A | V305E | -2.531 | Destabilizing | 0.179 | Increase |
| 28 | A | A308Y | -1.306 | Destabilizing | -0.725 | Decrease |
| 29 | A | C309Y | 0.844 | Stabilizing | -0.552 | Decrease |
| 30 | A | S310Y | 0.348 | Stabilizing | -0.630 | Decrease |
| 31 | A | D315E | -0.364 | Destabilizing | 0.331 | Increase |
| 32 | A | L384M | -1.266 | Destabilizing | -0.221 | Decrease |
| 33 | A | R392C | -0.347 | Destabilizing | 0.206 | Increase |
| 34 | A | P419S | 0.693 | Stabilizing | -0.661 | Decrease |
| 35 | A | R442Q | -0.180 | Destabilizing | 0.381 | Increase |
| 36 | A | P504S | -0.122 | Destabilizing | -0.052 | Decrease |
| 37 | A | P504L | 0.601 | Stabilizing | -0.050 | Decrease |
| 38 | A | I575L | -0.723 | Destabilizing | -0.013 | Decrease |
3.3. Effect of Nsp13 Mutation on Protein Function
The PROVEAN score predicts the functional impact of mutations in Nsp13. PROVEAN scores of mutants equal to or less than the default threshold score (-2.5) were predicted to have a deleterious effect on protein whereas PROVEAN scores more than the threshold score were predicted to have a neutral effect on the respective protein. A total of 38 mutations were reported in Nsp13 among Indian SARS-CoV-2 sequences, including 15 mutants as neutral and 23 mutants as deleterious based on their predicted PROVEAN scores, as mentioned in Table 3.
| S. No | Mutations | PROVEAN Score | Effect on Protein |
|---|---|---|---|
| 1 | P53S | -1.633 | Neutral |
| 2 | G54S | -4.046 | Deleterious |
| 3 | T58A | -0.506 | Neutral |
| 4 | M68K | -4.499 | Deleterious |
| 5 | V98F | -2.368 | Neutral |
| 6 | S100G | -3.933 | Deleterious |
| 7 | E142V | -6.883 | Deleterious |
| 8 | V154L | -0.076 | Neutral |
| 9 | H164Y | 1.312 | Neutral |
| 10 | S166A | -1.205 | Neutral |
| 11 | T214I | -5.162 | Deleterious |
| 12 | V226L | -2.763 | Deleterious |
| 13 | L227Q | -5.900 | Deleterious |
| 14 | T228K | -5.300 | Deleterious |
| 15 | S236I | -3.281 | Deleterious |
| 16 | A237T | -3.781 | Deleterious |
| 17 | H245R | 1.779 | Neutral |
| 18 | V247F | -1.795 | Neutral |
| 19 | Y253H | -1.608 | Neutral |
| 20 | S259T | -1.184 | Neutral |
| 21 | M274V | -1.983 | Neutral |
| 22 | H290Y | -5.300 | Deleterious |
| 23 | A296S | -1.750 | Neutral |
| 24 | L297F | -2.686 | Deleterious |
| 25 | P300L | -6.932 | Deleterious |
| 26 | I304K | -6.272 | Deleterious |
| 27 | V305E | -5.552 | Deleterious |
| 28 | A308Y | -5.900 | Deleterious |
| 29 | C309Y | -8.317 | Deleterious |
| 30 | S310Y | -5.786 | Deleterious |
| 31 | D315E | -3.933 | Deleterious |
| 32 | L384M | -1.983 | Neutral |
| 33 | R392C | -2.731 | Deleterious |
| 34 | P419S | -8.000 | Deleterious |
| 35 | R442Q | 0.467 | Neutral |
| 36 | P504S | -6.025 | Deleterious |
| 37 | P504L | -8.158 | Deleterious |
| 38 | I575L | -1.800 | Neutral |
3.4. Prediction of B-cell Epitopes
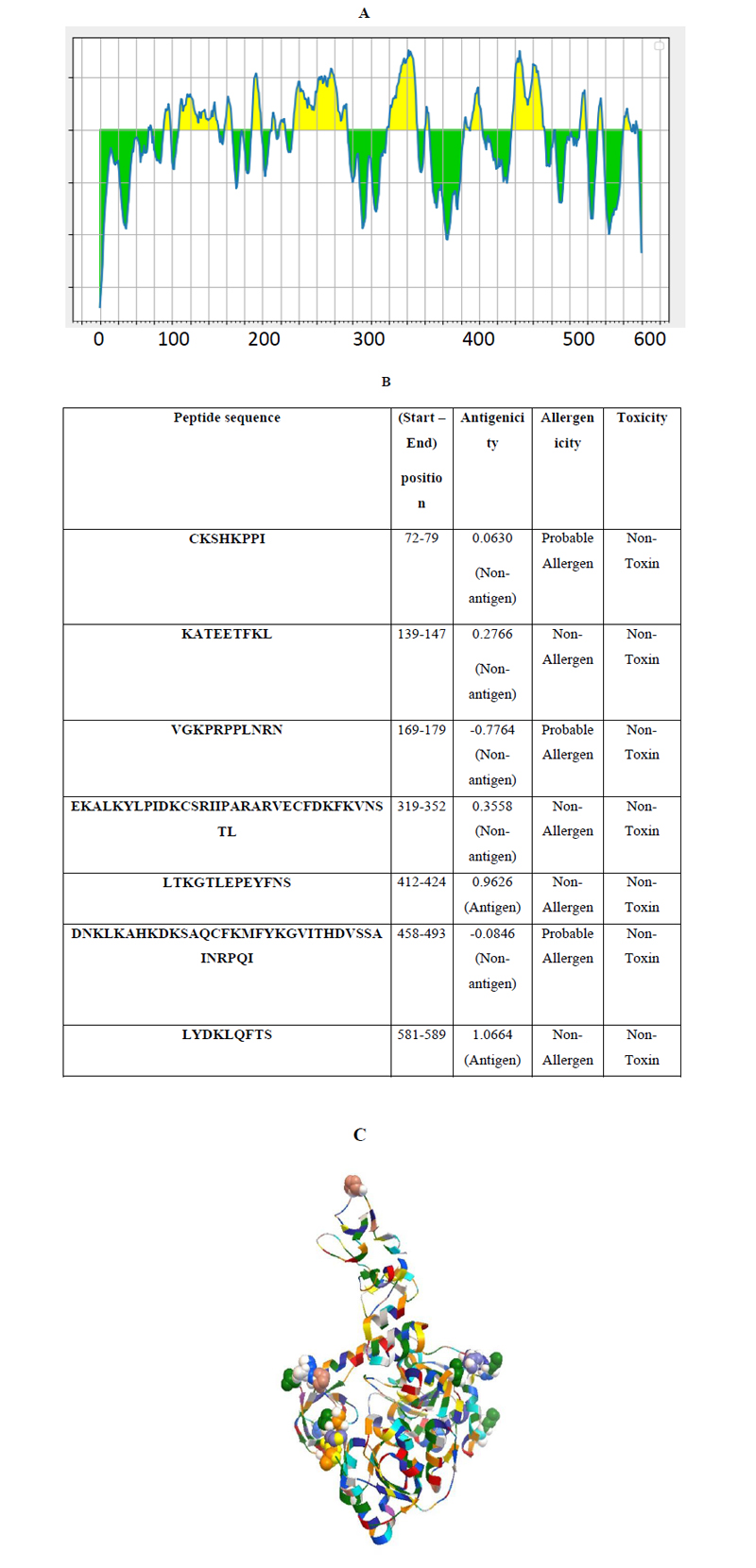
Linear continuous B-cell epitopes of SARS-CoV-2 Nsp13 were predicted by the IEDB web server tool, as shown in Fig. (3A). The BepiPred score of Nsp13 residues depicted a minimum score of -0.161 and a maximum score of -0.651 at a threshold value of 0.482. The data set identified the top seven linear epitopes with at least eight amino acid residues, of which both their sequences and locations are mentioned in Fig. (3B). These epitopes were further analyzed for antigenicity, allergenicity, and toxicity by in-silico based approaches. The predicted data showed that all of the seven peptides are non-toxic based on the Toxin Pred prediction tool. Out of the seven peptides, three peptides were identified as allergens and four as non-allergens based on allergenicity prediction (Fig. 3B). Similarly, two of these predicted linear epitopes exhibited antigenicity and the remaining five peptides were non-antigenic as analyzed by the VaxiJen prediction tool (Fig. 3B). In addition, discontinuous B-cell epitopes of Nsp13 were predicted using Discotope 2.0 server tool. The available 3D structure of Nsp13 in PDB format (PDB ID: 6ZSL) with a chain A of protein was used as input in the Discotope 2.0 server tool (Fig. 3C). The residue with Discotope score equal to or greater than the threshold value (-6.6) was identified as discontinuous epitope residues of Nsp13. The data sets identified 18 B cell epitope residues out of the total residues of Nsp13 protein at a threshold value of -6.6 (Fig. 3D). Histidine and valine at positions 245 and 247, out of 18 predicted residues were significantly positively predicted with high DiscoTope score values, as depicted in Fig. (2D). Overall, this data revealed B-cell epitopes contributed by Nsp13.
4. DISCUSSION
In the present study, we identified 38 mutations in Nsp13 among Indian isolates. The variations in the SARS-CoV-2 protein enable us to better understand its diversity, dynamics, and genetic epidemiology [31], which might provide an opportunity in developing effective and safe therapeutic drugs and vaccines for this highly mutable coronavirus. Our data revealed that the observed mutations at different positions (H164Y, A237T, T214I, C309Y, S236I, P419S, V305E, G54S, H290Y, P53S, A308Y, and A308Y) in the Nsp13 significantly affected the stability and flexibility of Nsp13 protein. In addition, the alterations in the function of Nsp13 were also predicted upon mutational effect by predicted PROVEAN scores. Furthermore, B-cell epitopes contributed by Nsp13 were identified using various predictive immunoinformatic tools. Immunological parameters of Nsp13, such as antigenicity, allergenicity, and toxicity were assessed to predict the potential B-cell epitopes. Predicted in silico epitopes are in consideration for the development of epitope-based peptide vaccine candidates against SARS-CoV-2. The predicted peptide sequences were correlated with the observed mutants. Our predicted data showed that there are seven high-rank linear epitopes as well as 18 discontinuous B-cell epitopes based on immunoinformatic findings. Moreover, it was investigated that, out of the total 38 identified mutations among Indian SARS-CoV-2 Nsp13 protein, four mutant residues at positions 142 (E142), 245 (H245), 247 (V247), and 419 (P419) were localised in the predicted B-cell epitopic region (Fig. 3). These mutant residues may help the SARS-CoV-2 variants in eliciting a distinct immune response from the wild-type SARS-CoV-2. Several previous studies evidently supported the findings with similar observations using the immunoinformatic approach [32-34]. Hence, our study provides some insights into understanding the Nsp13 epitopes, which could regulate host immune responses against SARS-CoV-2. The present in silico study revealed the impact of the identified mutations on the stability and flexibility of Nsp13 protein. Further in vivo studies are necessary to better understand and validate the effect of mutations on the immunogenicity of epitopes of SARS-CoV-2.

CONCLUSION
The emergence of a novel coronavirus, SARS-CoV-2, has become a major global concern with an unprecedented public health crisis. Understanding the genomic variations in SARS-CoV-2 could help in improving the current diagnostic techniques and developing suitable vaccine candidates against SARS-CoV-2 infections. Altogether, the results of the present in silico study showed various mutations in Nsp13 among Indian SARS-CoV-2 and their implications on the stability, flexibility, and function of Nsp13 protein using predictive tools of immunoinformatics. Non- structural proteins exhibited low glycation density compared to the structural proteins of SARS-CoV-2. Therefore, epitopes of Nsp13 could be used as effective and promising targets against SARS-CoV-2.
AUTHORS’ CONTRIBUTION
AK conceived the idea of the study and contributed to study design, data interpretation, and manuscript preparation. DK performed the experiments, analyzed the data, and prepared the manuscript. NK, SK, PKS, SKS, and NRB contributed to the conceptualization of the study and reviewing of the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No humans and animals were used in the studies.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declare none.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article.

