
Comparative Functional Genomics Studies for Understanding the Hypothetical Proteins in Mycobacterium Tuberculosis Variant Microti 12
Authors Info & Affiliations
Abstract
Background:
The Mycobacterium tuberculosis complex (MTBC) bacteria include the slowly growing, host-associated bacteria Mycobacterium tuberculosis, Mycobacterium Bovis, Mycobacterium microti, Mycobacterium africanum, Mycobacterium pinnipedii.
Aim:
Comparative Functional Genomics Studies for understanding the Hypothetical Proteins in Mycobacterium tuberculosis variant microti 12.
Objective:
A computational genomics study was performed to understand the 247 hypothetical protein genes. Functional annotation of virtual proteins was performed on different servers to maximize confidence level.
Methods:
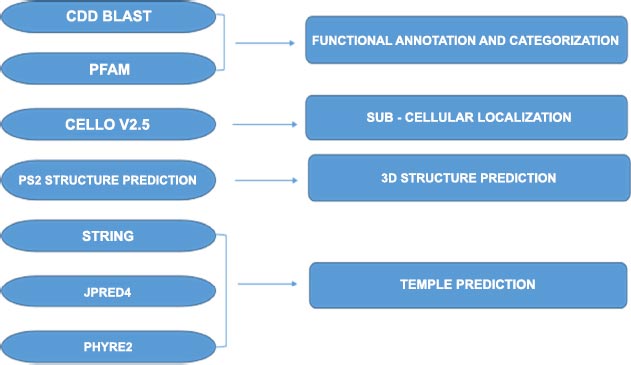
Sequence Retrieval. The whole genome sequences for the Mycobacterium tuberculosis micro variant 12 were retrieved from the KEGG database ( http://www.genome.jp/kegg/) and were used for screening 247 hypothetical proteins (Fig. 1). Functional Annotation and Sub-cellular localization. The Mycobacterium tuberculosis micro variant 12 hypothetical proteins were screened and sorted out from the genome and were individually analyzed for the presence of conserved functional domains by using computational biology tools like CDD-BLAST ( https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) ;Pfam ( http://pfam.xfam.org/ncbiseq/398365647); The subcellular localization of hypothetical proteins was determined by CELLO2GO ( http://cello.life.nctu.edu.tw). These web tools can search the defined conserved domains in the sequences available in the online servers or databases and assist in the classification of proteins in the appropriate families. Protein Structure Prediction. The in-silico structure predictions of the hypothetical protein sequences showing functional properties were carried out by using the PS2 Protein Structure Prediction Server ( http://www.ps2.life.nctu.edu.tw/). The online server helps to generate the 3D structures of the hypothetical proteins. The server accepts the sequences in FASTA format as a query to generate resultant proteins 3D structures. The structure determination is completely based on the conserved template regions detected during functional annotations. Protein-protein interaction through String database: The interaction of each hypothetical protein analyzed for functional characteristics was subjected to a protein-protein interaction server for the prediction of a possible functional role in interaction amongst the available known proteins ( https://string-db.org/). This information can help us to further validated the functional role of such hypothetical proteins and their possible role in the Mycobacterium Tuberculosis micro variant. Protein secondary structure prediction through JPred4: The secondary structure prediction of all the hypothetical proteins was determined through JPred4 ( http://www.compbio.dundee.ac.uk/jpred4/index.html) and served to identify the available secondary structures in the unknown hypothetical protein sequences. These further help us to understand the available templates in the uncharacterized protein sequences for the prediction of novel functions associated with these proteins. The predictions were further characterized by the Phyre2 server for structural modeling and prediction of templates based on comparative analysis based on conserved domains. Protein modeling, prediction, and analysis through Phyre2. The hypothetical proteins which were identified to have functional properties were further characterized by the Phyre2 server ( http://www.sbg.bio.ic.ac.uk/phyre2) for structural modeling and prediction of templates based on comparative analysis based on conserved domains.
Results:
A computational genomics study was performed to understand the 247 hypothetical protein genes Functional annotation of virtual proteins, and was performed on different servers to maximize confidence level. The functional prediction was performed by CDD-Blast and Pfam. The gene sequences of proteins have probably been successfully functionally annotated, characterized, and their subcellular localization and 3-D structural predictions have been predicted computationally. Online automated bioinformatics tools such as CDD-Blast, Pfam, CELLO2GO and PS2-Server were used for the structural and functional characterization of screened hypothetical proteins. The structure, function, and subcellular localization of a hypothetical protein from Mycobacterium tuberculosis variant microti 12 have been obtained and presented (Fig. 2). Also, the three-dimensional structure generated after using the template with the highest score was displayed as the template ID in the structure column of the respective hypothetical protein. However, as systems biology denies hypothetical protein functions, the structures of such proteins can be tested through biological processes and experiments, making them suitable for understanding their role in the life cycle, pathogenesis, and drug development. We can further explore these predictive possibilities in pharmaceuticals, and other clinically relevant studies. This study by HP helped find structure-function relationships in Mycobacterium tuberculosis variant microti 12 using a variety of bioinformatics tools. The string database made predictions about protein-protein interactions and the template helped us predict a hypothetical protein structure and even helped us find its 3D protein structure. Protein profiling can be performed on structures retrieved from these servers. This is useful for proteomics studies, including protein-protein interactions, protein expression of specific hypothetical proteins, and post-translational modifications of protein-coding genes. Further understanding of these hypothetical proteins can help us to know more about the Mycobacterium tuberculosis complex (MTBC) and may assist in Drugs and inhibitors against different pathogens within this complex.
Conclusion:
The all-inclusive bioinformatic study has helped to functionally elucidate 247 hypothetical proteins, which have resulted and made it easier to understand many functional proteins available in the Mycobacterium tuberculosis micro variant 12. The subcellular localization of the 247 sorted hypothetical proteins was also carried & which further helped us understand the localization of identified enzymes or proteins. We have successfully characterized the 247 unknown proteins of hypothetical protein sequences from Mycobacterium tuberculosis micro variant 12 to validate their structure and functions of the gene products. These predicted functions and three-dimensional structures may lead to establishing their role in the life cycle of the bacterium. This computationally generated data can also be further used for developing new protocols for new vaccines against Mycobacterium tuberculosis micro variant 12 that are essential for preventing infection, diseases, and transmission.
This complete result of Hypothetical Protein is needed for further studies of the whole genomic of the Mycobacterium Tuberculosis micro variant 12 for their function interpretation which further help in the understanding of its functions as well as structure.
Moreover, this interpretation would help us to study the evolution of Mycobacterium Tuberculosis micro variant 12 which further helps in the process of discovering the drugs to inhibit the causes of diseases.
1. INTRODUCTION
Tuberculosis (TB) is a highly contagious infectious disease that primarily affects the lungs [ 1]. The Mycobacterium tuberculosis complex (MTBC) species, which includes Mycobacterium africanum, M. bovis BCG, Mycobacterium bovis, Mycobacterium canettii, and Mycobacterium microti, are genetically related species of the TB-causing bacteria [ 1]. The Mycobacterium tuberculosis micro variant 12 is a particularly virulent strain that has been found to cause TB infections in the lungs, although there are relatively few reports on this strain to date [ 2]. Transmission of TB occurs when droplet nuclei containing M. tuberculosis are inhaled and the bacteria then spread in the respiratory tract. This can happen through activities such as sneezing, coughing, talking, singing, or laughing [ 2]. The size of the droplets is an important factor in the spread of respiratory diseases, with larger droplets typically settling on the ground and smaller droplets drying out, while middle-sized droplets can reach the trachea and potentially infect other organs such as the spine, kidney, and brain [ 3]. The droplets are typically transferred through the nasal tract or mouth [ 3]. Despite the sequencing of its genome in 1998, TB remains a serious global health problem and is a leading cause of mortality worldwide [ 4]. This is largely due to the chronic nature of the disease, which is characterized by slow growth and intracellular pathogenesis. The Mycobacterium tuberculosis micro variant 12, in particular, has the potential to cause severe and widespread outbreaks, underscoring the need for continued research and efforts to control and prevent the spread of TB [ 2].
1.1. Mycobacterium Tuberculosis Micro Variant 12 Classification
M. tuberculosis is a rod-shaped, strictly aerobic bacteria that are small in size and exhibits acid-fast properties [ 5-8]. It is known for its slow growth rate, which can make it difficult to treat and control. Despite this slow evolution rate, the emergence and spread of antibiotic resistance in M. tuberculosis pose an increasing threat to global public health [ 5]. The host-pathogen interaction between humans and M. tuberculosis is believed to have a genetic component [ 6], as a group of rare disorders called Mendelian susceptibility to mycobacterial diseases has been observed in a subset of individuals with a genetic defect that results in increased susceptibility to mycobacterial infection [ 7].
| Mycobacterium tuberculosis micro variant 12 | Kingdom: Bacteria
Subkingdom: Posibacteria Phylum: Actinobacteria Subclass: Actinobacteridae Order: Actinomycetales Suborder: Corynebacterineae Family: Mycbacteriaceae Genus: Mycobacterium Species: Mycobacterium tuberculosis |
One area of study that has gained attention in recent years is the identification and characterization of hypothetical proteins (HPs). HPs are proteins predicted to be expressed from open reading frames that make up a significant portion of the proteome in both prokaryotes and eukaryotes. Genomic studies have allowed researchers to explore the identification of many therapeutic targets by understanding the functions and interactions of these proteins [ 9]. Functional and structural predictions of hypothetical proteins may lead to the discovery of new functions and structures, as they can help us in many ways, such as pharmacological, markers targets, or drugs related to microorganisms, with the help of various methods [ 10].
This provides a further overview of the analytical techniques used to validate protein characterization It is also useful for protein expression, understanding protein-protein interactions, and the study of non-protein molecules that can regulate complex processes within cells. Ultimately, this will accelerate different areas of genomics focused on hypothetical or uncharacterized proteins. Describing HPs in specific genomes will help discover new structures and new functions that allow them to be further classified into different protein pathways. HP also supports drug design and discovery, markers, and pharmacological targets for discovery and screening. Investigating and describing the functions of HPs in pathogenic microbes that cause multiple diseases in humans and animals is of great importance to aid in linkage studies for drug discovery [ 9, 10].
Detection of HPs also aids in the discovery of previously unknown or unpredicted genes, which are very useful in genomics. Hypothetical proteins are unclassified proteins with unknown functions and in silico methods can be applied to model the structures of such hypothetical proteins and assist in designing experiments to identify their functions. Due to the majority of hypothetical proteins, pseudogenes, lacking functional activities has increased the interest in further study and understanding of unknown and unclassified proteins in various microorganisms including M. tuberculosis and their phages [ 11, 12].
Evolving online bioinformatics tools and servers have facilitated genome annotation to reveal the function of specific genes (proteins) and to determine the presence of enzymatically conserved domains within sequences to classify proteins into specific families and three-dimensional structures of protein sequences. There is particular interest in identifying and understanding the structural and functional properties attributed to hypothetical proteins present in Mycobacterium tuberculosis micro variant 12 [ 11, 12] Bioinformatics tools such as CDD-Blast and Pfam can predict functional properties of hypothetical proteins by comparing them to biological sequence databases. CELLO2GO determines the most likely subcellular localization, whereas protein structure prediction (PS2) helps predict the 3D structure of the target sequence.
Computer-assisted in silico studies of Mycobacterium tuberculosis microvariant 12 hypothetical proteins (uncharac- terized proteins) help predict the functional properties of these proteins using different bioinformatics tools and servers. This can aid in the discovery of new drug targets and markers for the development of new drugs to combat M. tuberculosis infections. Detection of HPs also aids in the discovery of previously unknown or unpredicted genes, which are very useful in genomics. In conclusion, the study of hypothetical proteins in M. tuberculosis is of great importance in understanding the mechanisms of the pathogenicity of this microorganism, and in the discovery of new targets for the development of new drugs.

2. MATERIALS AND METHODS
2.1. Sequence Retrieval
In this study, we utilized the KEGG database ( http://www.genome.jp/kegg/) [ 1, 13, 14], which is a widely accepted source for genomic data, to obtain the genome sequences of the Mycobacterium tuberculosis micro variant 12. We then used these sequences to screen a total of 247 hypothetical proteins using the information and tools provided by KEGG. The results of the screening process are presented in Fig. ( 1). We aimed to conduct an in-depth analysis of the M. tuberculosis microvariant 12 genome to identify potential targets for the development of new treatments and therapies for tuberculosis.
2.2. Functional Annotation and Sub-cellular Localization
The Mycobacterium tuberculosis micro variant 12 genome was analyzed in order to identify and screen hypothetical proteins. These proteins were then individually examined for the presence of conserved functional domains using computational biology tools. The tools used in this analysis include CDD-BLAST ( https://www.ncbi.nlm.nih.gov/ Structur e/cdd/wrpsb.cgi) [ 15, 16], Pfam ( http://pfam.xfam.org/ncbiseq /398365647) [ 17] and CELLO2GO ( http://cello.life.nctu.ed u.tw) [ 18]. These tools are widely used in the field of computational biology and are designed to search for defined conserved domains in the sequences available in online servers or databases. This analysis enabled the classification of proteins in the appropriate families and provided valuable information about the subcellular localization of the hypothetical proteins. The use of these web-based tools greatly facilitated the analysis process and allowed for more efficient identification of potential targets for the development of new treatments and therapies for tuberculosis.
2.3. Protein Structure Prediction
The functional properties of the hypothetical protein sequences identified from the Mycobacterium tuberculosis micro variant 12 genome were further characterized through in-silico structure predictions. This was achieved by using the PS2 Protein Structure Prediction Server ( http://www.ps2. life.nctu.edu.tw/), which is a widely-used and respected resource for predicting the 3D structures of proteins [ 19]. The PS2 server accepts the protein sequences in FASTA format as a query and utilizes computational techniques to generate 3D structures of the hypothetical proteins based on the conserved template regions detected during functional annotations. This process allows for a more detailed understanding of the functional properties of the hypothetical proteins and can provide valuable information for the development of new treatments and therapies for tuberculosis. The PS2 server is widely used by researchers to predict the 3D structure of proteins and it has good accuracy and reliability.
2.4. Protein-protein Interaction through String Database
After analyzing the functional properties of the hypothetical proteins from the Mycobacterium tuberculosis microvariant 12 genome, the next step was to investigate their potential interactions with other known proteins. This was done by subjecting each protein to a protein-protein interaction prediction server, such as STRING ( https://string-db.org/), which is widely used in the field of bioinformatics [ 20, 21]. This server utilizes a variety of computational techniques to predict potential interactions between proteins based on their amino acid sequences, structures, and functional domains. The information obtained from this analysis can provide valuable insights into the functional roles of the hypothetical proteins and their potential contribution to the biology of the Mycobacterium tuberculosis microvariant 12. By using STRING, we can predict the functional interactions between the proteins, and validate the functional role of such hypothetical proteins and their possible role in the Mycobacterium Tuberculosis micro variant pathogenesis.
2.5. Protein Secondary Structure Prediction through JPred4
The secondary structure of all hypothetical proteins was determined using the JPred4 program ( http://www.compbio. dundee.ac.uk/jpred4/index.html). This method allows for the identification of the various secondary structures present within unknown hypothetical protein sequences [ 22]. This information is crucial for understanding the potential templates that can be used to predict novel functions associated with these proteins. To further analyze these predictions, we used the Phyre2 server, which employs comparative analysis based on conserved domains to perform structural modeling and template prediction. This combination of methods provides a comprehensive approach to understanding the potential functions of hypothetical proteins.
2.6. Protein Modeling, Prediction, and Analysis through Phyre2
The hypothetical proteins that were found to possess functional properties were further analyzed using the Phyre2 server ( http://www.sbg.bio.ic.ac.uk/phyre2) [ 23]. This powerful tool utilizes comparative analysis based on conserved domains to perform structural modeling and template prediction. By using this approach, we were able to gain a deeper understanding of the potential functions of these hypothetical proteins. The Phyre2 server allows for the identification of structural templates that can be used to predict the potential functions of these proteins, providing valuable insights into their potential biological roles. Furthermore, the Phyre2 server also allows us to predict the structural and functional characteristics of these proteins, which can be crucial for understanding the molecular mechanisms underlying their functions. Overall, the Phyre2 server is an important tool for the functional characterization of hypothetical proteins [ 24].
3. RESULTS AND DISCUSSION
A computational genomics study was performed to understand the 247 hypothetical protein genes Functional annotation of virtual proteins was performed on different servers to maximize confidence level. Computational genomics studies were aimed at understanding the functional annotation and subcellular localization of hypothetical proteins from Mycobacterium tuberculosis variant microti 12. The study involved the use of various bioinformatics tools, including CDD-Blast, Pfam, CELLO2GO, PS2-Server, Jpred 4, and PHYRE2. These tools were used to predict the structure and function of the screened hypothetical proteins.
CDD-Blast and Pfam were used for functional prediction by searching for conserved protein domains and motifs within the hypothetical proteins. This helped to provide insights into the potential functions of these proteins. CELLO2GO and PS2-Server were used to predict the subcellular localization of the hypothetical proteins. This information is important as it provides insights into the potential roles of the proteins within the cell.
Jpred 4 was used to predict the secondary structure of the proteins. This tool uses a combination of machine learning techniques and neural networks to predict the regular local patterns of folds that appear in a protein's backbone chain. The predicted secondary structure can be used to understand the protein's function and interaction with other molecules.
PHYRE2, on the other hand, was used to predict the 3D structure of the proteins using a template-based approach. This tool searches a database of known protein structures to find a template structure that is like the target protein. Once a suitable template is found, PHYRE2 aligns the target sequence to the template structure and uses this alignment to build a 3D model of the target protein. The accuracy of the prediction depends on the availability of a suitable template structure in the database.
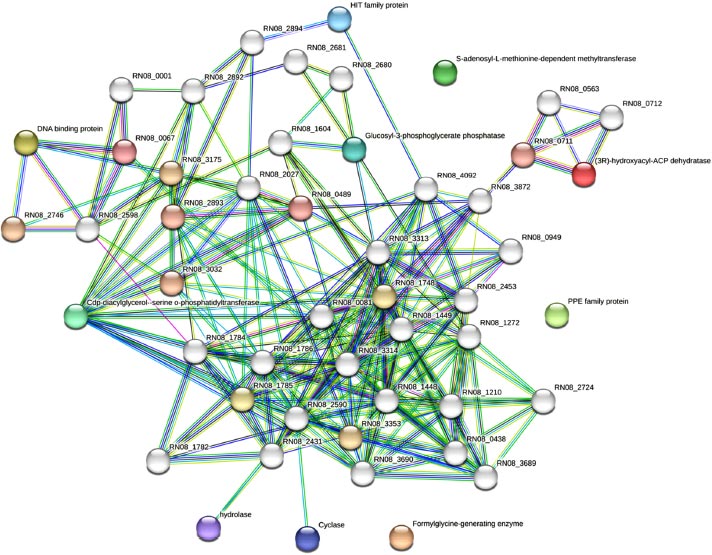
The study also used the STRING database to make predictions about protein-protein interactions. These predictions can help to provide insights into the potential roles of the hypothetical proteins within the cell as represented in Fig. ( 3). Detailed explanations are provided to support the predicted hypothetical proteins and their interactions determined through databases, text mining as well as experimentally reported studies in Table S1 and functional annotations in Table S2. Similarly, phylogenetic co-occurrences, co-expressions, as well as the chromosomal neighborhood were also determined by comparing all the predicted hypothetical proteins with Mycobacterium tuber- culosis variant microti organisms through string multiple protein search. The hydrolase was amongst the most frequently predicted hypothetical proteins followed by 2-aminomuconic 6-semialdehyde dehydrogenase, Monomethylamine methyl- transferase, Cyclase, CDP-diacylglycerol-serine o-phospha- tidyltransferase amongst many others. The servers used for predicting proteins have revealed that there are strong protein-protein interactions among the proteins, as depicted in Fig. ( 3). Interestingly, some of the hypothetical proteins identified were not found to be associated with any of the interacting proteins. This observation suggests the possibility of a novel metabolic pathway involving these proteins, which has not been explored in these strains before. Therefore, further in-depth research is required to better understand the role of these hypothetical proteins in prokaryotes and their potential importance in these organisms. By investigating these proteins, we may uncover novel biochemical pathways and functions that can shed new light on the metabolism and cellular processes of these microorganisms.
The results of the study revealed the structure-function relationships of the hypothetical proteins and provided insights into their potential roles in the Mycobacterium tuberculosis complex. The study highlights the usefulness of bioinformatics tools in predicting the structure and function of proteins, particularly hypothetical proteins whose functions are not yet known. However, it is important to note that while bioinformatics tools are useful in predicting protein structures and functions, these predictions need to be validated through biological experiments. This is particularly important for hypothetical proteins, whose functions are not yet known.
In conclusion, the study demonstrates the usefulness of bioinformatics tools such as CDD-Blast, Pfam, CELLO2GO, PS2-Server, Jpred 4, and PHYRE2 in predicting the structure and function of proteins. Similar studies have been reported for understanding hypothetical and unknown proteins in different microbial species [ 24]. These tools can be used to provide insights into the potential roles of hypothetical proteins in biological systems. However, it is important to validate these predictions through biological experiments. The study highlights the potential of bioinformatics in understanding the Mycobacterium tuberculosis complex and in developing drugs and inhibitors against different pathogens within this complex.
CONCLUSION
Our all-inclusive bioinformatic study has provided a wealth of information about the functions, structures, and subcellular localization of 247 hypothetical proteins from Mycobacterium tuberculosis microvariant 12. This information is crucial for understanding the mechanisms by which these proteins function within the bacterium, how they contribute to its life cycle, and how they may be targeted for the development of new vaccines and drugs.
The functional annotation of these hypothetical proteins provides a deeper understanding of the functions and structures of this bacterium, which can further aid in the study of its evolution. Additionally, this information can be used to develop drugs and inhibitors against the causes of diseases caused by this micro variant. The prediction of protein-protein interactions and the 3D structure of these hypothetical proteins can be useful for future studies of proteomics and drug development. Furthermore, the information we have generated through our study can be used as a guide for experimental studies.
Overall, this study highlights the importance of bioinformatic analysis in advancing our understanding of bacterial pathogens and their associated diseases. By leveraging computational methods and tools, we were able to provide a comprehensive characterization of the hypothetical proteins in Mycobacterium tuberculosis micro variant 12, which can guide further experimental studies and help prioritize which proteins should be targeted for drug and vaccine development.
Furthermore, the results of this study shed light on the role of these hypothetical proteins in the life cycle of Mycobacterium tuberculosis microvariant 12. By determining their subcellular localization and predicting their interactions with other proteins, we gained insights into their potential mechanisms of action and how they contribute to the bacterium's survival and virulence. Although our findings provide a promising starting point for future research, several limitations to this study should be acknowledged. First, our predictions are based solely on computational analysis and have yet to be confirmed experimentally. Therefore, caution should be exercised when interpreting the results, and further validation is required to confirm the functions and structures of these hypothetical proteins. Second, this study focused exclusively on Mycobacterium tuberculosis microvariant 12, and it is unclear whether the findings can be generalized to other bacterial strains or species. Future studies could explore whether similar bioinformatic methods can be applied to other pathogens to provide insights into their hypothetical proteins. Finally, this study only scratches the surface of the vast complexity of the Mycobacterium tuberculosis micro variant 12 proteome. As more data becomes available, it will be important to revisit and refine our predictions to gain a deeper understanding of the functions and structures of these proteins.
In conclusion, this study provides a valuable contribution to the field of bioinformatics and bacterial pathogenesis research. By characterizing the functions, structures, and subcellular localization of hypothetical proteins in Mycobacterium tuberculosis micro variant 12, we gained insights into their potential roles in the bacterium's life cycle and how they may be targeted for the development of vaccines and drugs. These findings lay the foundation for future studies and represent a step towards a better understanding of bacterial pathogens and their associated diseases.
AUTHOR'S CONTRIBUTIONS
Conceptualization, S.G.S.; methodology, T.V.S, T.G.S., and S.G.S.; software, T.V.S, T.G.S., and S.G.S.; validation, T.V.S, T.G.S., and S.G.S.; formal analysis, T.V.S, T.G.S., and S.G.S.; investigation, T.V.S, T.G.S., and S.G.S.; resources, V.V.C., A.R.G., G.V.U., A.S.J., and B.S.P.; data curation, T.V.S, T.G.S., V.V.C., A.R.G., G.V.U., A.S.J., B.S.P., S.G.S.; writing—original draft preparation, S.G.S.; writing—review and editing, T.V.S, T.G.S., V.V.C., A.R.G., G.V.U., A.S.J., B.S.P., S.G.S.; visualization, T.V.S, T.G.S., and S.G.S.; supervision, V.V.C., A.R.G., G.V.U., A.S.J., B.S.P., S.G.S.; project administration, S.G.S.; funding acquisition, S.G.S. All authors have read and agreed to the published version of the manuscript.”
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No humans or animals were used for the studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS:
All the data can be accessed at the respective servers or repositories cited in the article. The results generated are curated for publication purposes. No modification, manipulation, and falsification of data is reported through these studies.
FUNDING
This research received no external funding.
CONFLICT OF INTEREST
The authors declare no conflict of interest financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.
SUPPLEMENTARY MATERIALS
Supplementary material is available on the Publisher’s website.

